Abstract
Improvements in hemophilia care and antiviral treatments have resulted in increases in median life expectancy for persons with congenital hemophilia A and B. Currently, 2% of hemophilia A and B patients surveyed in US comprehensive hemophilia treatment centers are 65 years of age or older and 15% are 45 years or older. Many of the complications of hemophilia, including intracranial hemorrhage, joint disease, and inhibitor development, increase with increasing age. Hepatocellular carcinoma and end-stage liver disease are increasing in the older hemophilia population due to infection with hepatitis C (HCV) and HCV/HIV coinfection. Older hemophilia patients also now face the same medical conditions associated with aging in the general population, including cardiovascular disease and cancer. Complex hemostatic management, sometimes in conjunction with antithrombotic management, with extensive cross-specialty clinical and laboratory coordination may be required for the care of the older hemophilia patient. Because elderly hemophilia patients currently represent a small portion of the overall hemophilia population, there is little in the way of clinical data to guide recommendations. Registry databases and cooperative group studies are needed for the development of evidence-based guidelines for the older hemophilia population, which is anticipated to expand in the future.
Introduction
Advances in coagulation protein replacement therapy, the development of specialized comprehensive care centers, and utilization of home therapy and factor prophylaxis have led to progressive reductions in morbidity and increases in life expectancy for persons with congenital hemophilia A and B.1–4 Before the 1970s, with only the availability of cryoprecipitate and low-purity plasma fractions enriched with factor VIII, patients with hemophilia had a significantly reduced life expectancy, with intracranial or other hemorrhages the major cause of mortality.5 Median life expectancy in males with severe hemophilia was 11 years in the early 20th century, and had increased to the range of 55 to 63 years in the 1970s, still a reduced life span compared with the overall male population.5,6
Plasma-derived factor concentrate replacement therapy became commercially available in the United States and Europe in the early 1970s, and by 1982, 73% of persons with hemophilia A in the United States received factor concentrates, increasing the median life expectancy by the early 1980s to almost 68 years.7,8 Specialized hemophilia treatment centers were established in the mid-1970s, which improved medical management of hemophilia, but hemorrhage still remained the major cause of death in hemophiliacs in 1982.7 That same year, the first cases of HIV were identified in the hemophilia population,9 and by 1990, the median life expectancy among US hemophiliacs had dropped to 49 years and the mean age at death to 40 years.7 Clotting factor concentrates virally safe from HIV have been available since 1985, and hepatitis B and C virally safe concentrates have been available since 1992.10 Viral safety was achieved initially using viral-inactivation methods for plasma-derived factor concentrates before recombinant factor products were developed. By 2001, hemophilic (all severities combined) life expectancy in The Netherlands had increased to 67 years (74 years excluding virally infected hemophiliacs), and by 2007, the overall hemophilic life expectancy was reported to be 71 years in Italy.5,11 In the United States, the utilization of specialized comprehensive care centers resulted in a 30% decrease in mortality for males with hemophilia compared with those in other sources of care.2
Males with hemophilia who are age 45 and older, meaning that they were born in 1965 or earlier, prior to widely available factor concentrates and prior to virally safe factor concentrate products, are the subject of this section. The complications, comorbidities, and management issues confronting the clinician taking care of the older male hemophilia A or B patient are presented. Some of the management issues unique to the older hemophilia patient pose particular challenges to the clinician, with limited clinical data available to guide recommendations.
Characteristics of the Older Hemophilia Population
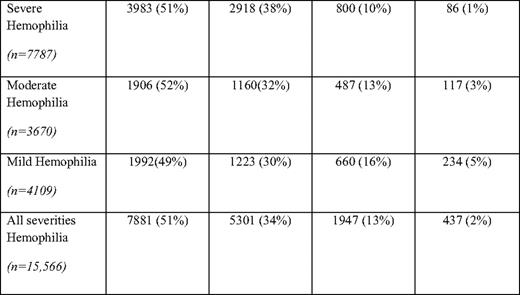
Based on surveillance data from comprehensive hemophilia treatment centers, currently in the United States, individuals 45 years and older with hemophilia A and B comprise 15% of the total hemophilia population, with those older than 65 years comprising 2% of the total hemophilia A and B population (Table 1). Five percent of mild (> 5 U/dL factor VIII or IX) hemophilia patients are over 65 years old, whereas only 1% of severe (< 1 U/dL factor VIII or IX) hemophilia patients are over 65 (Table 1). These figures contrast with the proportion of US males over 65 years (10%) and those over 45 years old (32%) (according to the 2000 US Census). The percentage of persons with hemophilia age 65 and older in Italy has been reported to be 6.4%,12 higher than the percentage observed in the United States.
Viral Disease
With the advent of highly active antiretroviral therapy (HAART), the survival of HIV-infected individuals with hemophilia has improved significantly, with 27% to 39% surviving 20 to 25 years.13–15 Younger age at the time of HIV seroconversion has been associated with improved survival.14,16 Most HIV-infected hemophilia patients are coinfected; among 458 HIV-positive hemophilia A and B patients in Canada, 96.5% were hepatitis C (HCV) positive.14 Both cumulative incidence of end-stage liver disease and fatality due to liver failure have been reported to be significantly increased in the dually infected hemophiliac compared with the HCV-monoinfected hemophiliac.15,17 HAART was associated with an increased time to end-stage liver disease in HCV/HIV positive patients, similar to that observed in HCV positive, HIV negative patients.18
Hepatitis B and C markers are highly prevalent among older males with hemophilia. Between 85% and 93% of US males with hemophilia born before 1975 have been found to be positive for HCV infection, and 27% to 51% of similar birth cohorts have been found to be positive for hepatitis B infection.10 A significant difference between HCV-infected hemophilia patients and non-hemophilia patients is the duration of their infection. Hemophilia patients usually acquired their HCV infection in the first year of life with their first factor infusion, and pre-liver transplant survival in hemophilia patients has been reported to be much shorter than in non-hemophilia patients.19 HCV antiviral treatment consists of therapy with pegylated interferon and ribavirin, which achieves a 50% response in hemophilic patients with genotype 1 (most common) or genotype 4, compared with an 80% to 90% response in genotypes 2 and 3. Coinfection with HIV also adversely affects the effectiveness of treatment.20 If an assessment of fibrosis is needed for determination of treatment, a transjugular liver biopsy with adequate factor coverage is preferred over a percutaneous biopsy to reduce the risk of bleeding. Factor VIII or IX replacement to achieve levels 80 to 100 U/dL prior to biopsy, with continued factor replacement to maintain levels >50 U/dL for 2 to 4 d following biopsy, has been associated with a 1% to 2% rate of clinically significant bleeding.21 Recently, transient elastography has been proposed as a noninvasive tool for the assessment of liver fibrosis in hemophilia patients.22 Liver transplantation is an option for hemophilia patients with end-stage liver disease who are infected with HCV or HCV-/HIV and corrects the factor deficiency.
In hemophilia patients 65 years and older who received replacement products prior to viral-inactivation techniques, viral infection is almost universal. Among 39 Italian patients 65 years of age or older with severe hemophilia A and B, 38 had blood-borne viral infections, including 13% with HIV, 92% with HCV, and 10% with hepatitis B.12 While death from HIV-related causes has decreased significantly, hepatitis and other liver-related deaths have increased, and the management of hepatitis, chronic liver disease, liver failure, and hepatocellular carcinoma represent major management challenges in the older hemophiliac. Management of bleeding complications in those with thrombocytopenia secondary to chronic liver disease and/or hepatitis may entail factor replacement to normalize levels.
Intracranial Hemorrhage
Hemorrhage is the third leading cause of mortality in hemophiliacs after HIV and hepatitis, with intracranial hemorrhage constituting 30% to 50% of hemorrhagic deaths.5,14,23 Data from the six-state Hemophilia Surveillance System Project cohort in the United States demonstrated that age greater than 51 years, severe hemophilia, HIV, and the presence of inhibitor were independent risk factors for intracranial hemorrhage.24 An effect of inhibitor presence on intracranial hemorrhage-associated mortality, however, was not confirmed by UK hemophilia surveillance data.25 Intracranial hemorrhage has the highest mortality of any hemophilia-related bleeding, and a death rate of 30% has been reported among hemophilia patients age 50 and older with intracranial hemorrhage.25 The majority of intracranial hemorrhages in this population are non-traumatic. Intracranial hemorrhage typically presents with headache, and immediate factor infusion sufficient to raise factor VIII or IX levels to 100 U/dL prior to diagnostic evaluation with a non-contrast computed tomography (CT) scan is important. If confirmed, factor infusion sufficient to maintain a trough of more than 50 U/dL is recommended for at least 10 to 14 d.26,27 Laboratory monitoring of factor VIII or IX levels before and after factor infusion is suggested in order to adjust the dose of factor concentrate to maintain adequate levels. The off-label use of antifibrinolytics as adjunctive management has been used, but efficacy data are lacking. Hemophilia patients who have had an intracranial hemorrhage are candidates for long-term prophylaxis with factor concentrates or prophylaxis for 2 years at a minimum.26,27
Joint Disease
The US Joint Outcome study demonstrated the benefits of factor concentrate prophylaxis in preventing joint damage and bleeding episodes compared with on-demand factor use in children.4 Individuals age 65 and older with hemophilia did not have access to regular replacement therapy until well into adulthood, and those 45 and older did not have ready access during significant portions of their childhood. Joint disease remains a major cause of morbidity in the older hemophilia patient. The majority of severe hemophilia patients age 65 and older have arthropathy in four to six of the six joints most commonly affected by bleeding (i.e., knees, ankles, and elbows).12,28 Arthropathy is also associated with reduced bone mineral density in severe hemophilia. Pain management includes acetaminophen, cyclooxygenase 2 (COX2) inhibitors, and narcotics.29 Managing painful arthropathy with nonaddictive medications is difficult in the older hemophilia patient because of increased risks of nonsteroidal anti-inflammatory agent (NSAID)-associated gastrointestinal bleeding and acetaminophen-associated liver dysfunction.30,31 In one study, the use of nonselective NSAIDs was associated with an increased likelihood of bleeding, whereas selective COX2 inhibitors were not.30 The extent of use of long-term continued factor prophylaxis initiated after multiple joint bleeds and established joint damage, so-called “secondary” prophylaxis, in adults over 45 is unknown; its effectiveness in mitigating hemophilic arthropathy is also unknown. A European survey found that 23% (58 out of 251) of severe hemophiliacs over 50 years of age were on a regimen of regular concentrate administration,32 but this may not be similar in the United States, given the variation between the United States and some European countries in prophylaxis practice patterns for pediatric hemophilia patients. Orthopedic surgical procedures, including ankle arthrodesis and hip and knee arthroplasty, are frequently performed to reduce the pain and disability of hemophilic arthropathy when conservative management fails.
Hemostatic management of the older severe hemophilia patient undergoing major orthopedic surgery is challenging, and should take into account inhibitor status and risk of venous thromboembolism.33 In a literature review of replacement therapy for invasive procedures, 35 clinical studies, which included 707 orthopedic surgeries, were identified.34 Sixteen percent of major surgical procedures were managed by continuous infusion of factor concentrate (off-label) and the rest by bolus infusion. The preoperative target factor VIII or IX level was > 80 U/dL in 26 out of 31 studies, and among 27 studies addressing postoperative trough levels, 19 out of 27 targeted troughs of > 50 U/dL in the first postoperative week. The majority of studies targeted a trough level of > 30 U/dL in the second postoperative week. Factor concentrate may need to be continued beyond the second postoperative week prior to physical therapy sessions. Preoperative pharmacokinetic evaluation for determination of factor recovery and half-life is performed in many hemophilia treatment centers34 for planning perioperative hemostatic management, and the availability of factor VIII or IX laboratory assessment during the postoperative period is important for factor concentrate adjustment. Hemostatic management of hemophilia patients with inhibitors who are undergoing major surgery involves the extensive use of bypassing agents.33 The risk of venous thromboembolism in older hemophiliacs undergoing major orthopedic surgery is not well established. Furthermore, the impact that elevated factor VIII levels due to excessive factor concentrate replacement have on thrombosis risk in this population is not known. The use of anticoagulant thromboprophylaxis following major orthopedic surgery in the patient with hemophilia is controversial. Some advocate the use of prophylactic low-molecular-weight heparin in the hemophiliac without an inhibitor,33 but this should only be considered during periods of factor concentrate replacement with close factor level monitoring and only when trough factor VIII or IX levels are within the normal or near-normal range. Mechanical methods of thromboprophylaxis and early ambulation are otherwise recommended.
Inhibitors
Cumulative inhibitor risk increases with age.25 According to UK surveillance data, the cumulative risk of hemophilia A inhibitor at 15 years of age is 20%, at 50 years 30%, and at 75 years 36%. In moderate and mild hemophilia A, the cumulative risk is 6%, 10%, and 12% at 15, 50, and 75 years of age, respectively. For hemophilia B, the cumulative risk is much lower at age 75 (8%) and at all ages compared with hemophilia A.25 Older individuals with mild hemophilia A may be at increased risk for inhibitor development when receiving intensive perioperative factor concentrates, and should be monitored for the presence of inhibitor subsequent to factor exposure.28,35 The risk of inhibitor development in mild and moderate hemophilia A has been reported to be higher in patients exposed to continuous infusion and in those with certain missense mutations, especially Arg531Cys.35 Hemophilia patients emigrating from countries where factor concentrates are not readily available may be at increased risk for inhibitor development when first exposed to factor concentrates in the United States.
The management of inhibitors in the older hemophilia patient poses challenges, not only because of the increased risk of bleeding, but also because of the potential increase in thrombotic risk with the use of bypassing agents. The role of immune tolerance induction for inhibitor eradication in this age group is not known. A recent report analyzing the use of rituximab among cases reported in the literature, which included 9 out of 46 patients over age 50, demonstrated an increased response to rituximab with age and with mild/moderate hemophilia.36
Cardiovascular Disease
Hemophilia seems to protect against cardiovascular disease.5,6,37,38 Several European studies have documented a 38% to 80% decreased mortality from ischemic heart disease in males with hemophilia compared with the non-hemophilia male population.5,6,38 One US study, however, reported an increased mortality for acute myocardial infarction in hemophilia patients compared with the general US male population.2 Based on hospital discharge data from six US states from 1993 to 1998, ischemic heart disease was found to be reduced in hemophilic males ages 45 to 64 and those 65 and older compared with normal US males.37 It has been postulated that hypocoagulability protects against thrombus formation in the hemophiliac, but whether it also protects against atherosclerosis is not clear. In hemophilia patients, age, hypertension, diabetes, and hyperlipidemia have been independently associated with the risk of ischemic heart disease.37 However, a study comparing hemophilic patients with age-, gender-, and race-matched controls demonstrated no differences in intraluminal coronary artery stenosis at autopsy, or in cardiovascular risk factors including hypertension, hypercholesterolemia, smoking, or diabetes.39 Routine cardiovascular preventive care and risk-reduction measures are indicated in individuals with hemophilia29,40
HIV is an additional risk factor that has been found to be independently associated with non-ischemic heart disease in males with hemophilia.37 A recent study in non-hemophilic HIV patients demonstrated that diastolic dysfunction and left ventricular mass were independently associated with HIV infection.41 Other explanations for an increased cardiovascular risk with HIV include the association of some antiretrovirals with dyslipidimia (and independently with cardiovascular risk) and the association of HIV with inflammation and immune activation.42 These risks may be important in the hemophilia population over 45 years of age given the prevalence of HIV in this group.
Managing cardiovascular disease and associated risks in the hemophilia patient is a challenge, and there are few clinical studies beyond case reports or small series to guide treatment. In one case series, cardiac surgery in six hemophilia A patients, including four mild, one moderate, and one severe hemophiliac, undergoing aortic valve replacement and/or coronary artery bypass graft with extracorporeal circulation were managed with recombinant factor VIII replacement for between 11 and 24 d. All valve replacements were tissue valves to avoid the need for long-term anticoagulation. All received factor replacement by bolus infusion, 4 out of 6 received antifibrinolytics postoperatively, and 4 out of 6 received low-molecular-weight heparin for up to 3 months postoperatively. Postoperative morbidity was reported to be similar to patients without hemophilia.43 Some have suggested using continuous infusion (off-label) recombinant factor VIII or IX over bolus factor infusion perioperatively to avoid peaks of factor VIII or IX activity, to maintain stable factor levels, and to reduce thrombosis risk.29,40 Because of the bleeding risks in patients with hemophilia, cardiac bypass surgery may be preferred over percutaneous coronary intervention with stenting given the requirement for dual antiplatelet agents for extended periods of time, the risk of re-stenosis, and the potential need for repeated procedures.29 Bare metal stents are preferred over drug-eluting stents because of the considerably shorter duration of antiplatelet therapy required with bare metal stents. Factor replacement to maintain a trough plasma factor level of at least 30 U/dL for the duration of the antiplatelet therapy has been recommended.33,44 Close laboratory monitoring of factor levels periprocedurally, perioperatively, and during the use of anticoagulants and/or antiplatelet agents is required.
Long-term antithrombotic therapy for the management of coronary artery disease or atrial fibrillation in persons with hemophilia is largely based on opinion, because risk-stratification models have not yet been determined in this population.33,44,45 The long-term use of low-dose aspirin in patients with mild or moderate hemophilia has been proposed, but extended use in patients with severe hemophilia may be difficult, even with factor prophylaxis and close monitoring for bleeding.44 The use of warfarin has generally not been recommended.
Cancer
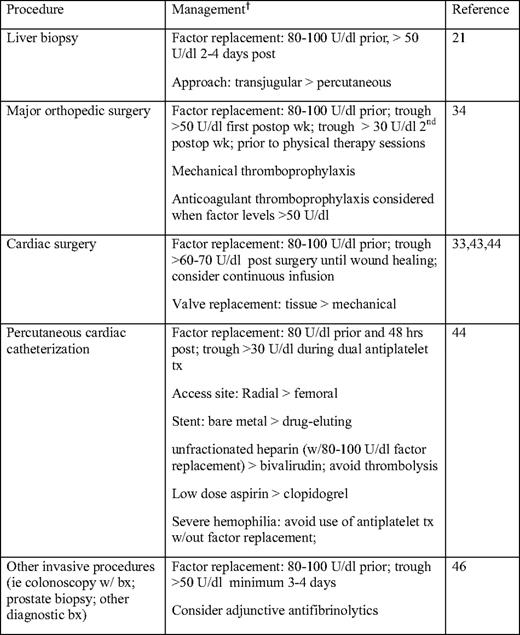
Older hemophilia patients are at increased risk for HCV-associated hepatocellular carcinoma and HIV-associated non-Hodgkin's lymphoma, despite substantial reductions in HIV-associated malignancies with HAART.5,46 Hepatocellular carcinoma is an increasingly important cause of mortality, with a reported standardized mortality ratio of 17.2 (95% confidence interval 5.2–35.9).5 Age over 45 years, older age at the time of HCV infection, elevated alpha fetoprotein, and the presence of cirrhosis are risk factors for hepatocellular carcinoma.47 Regular surveillance with alpha fetoprotein levels and ultrasonography and/or contrast-enhanced imaging has been recommended in this age group, particularly in individuals with cirrhosis, in order to improve detection of earlier-stage hepatocellular carcinoma.29,48 Most studies have not found an increased incidence of other malignancies in hemophiliacs compared with the general population, but the studies have not been uniform.45,46 The elderly hemophiliac with cancer should be managed similarly to the non-coagulopathic individual; however, factor replacement is needed for invasive procedures, and prophylactic factor replacement may be indicated when platelet counts are low.33,45 Just as in the general population, age-appropriate screening for malignancies such as colon and prostate cancer is indicated in patients with hemophilia, with prophylactic factor replacement for invasive screening procedures (Table 2).
Conclusions
Many of the complications of hemophilia, including inhibitor development, intracranial hemorrhage, and joint disease, are increased with increasing age. Viral infection with HIV and HCV continue to play dominant roles in the older hemophilia population, with significant increases in HCV-associated hepatocellular carcinoma and liver failure and increasing evidence for a role of HIV in cardiovascular disease in this population. Age-related comorbidities in hemophilia patients may require complex treatment and extensive cross-specialty clinical and laboratory coordination. With little in the way of evidence-based guidelines, the management of older patients with hemophilia remains a challenge for hematologists. Registry databases and collaborative studies are needed to establish evidence-based guidelines for the care of this emerging population.
Disclosure
Conflict-of-interest disclosure: The author has received research funding from Baxter, Pfizer, Octapharma, Bayer, and CSL Behring. The author has received an honorarium and been a member of an advisory board for CSL Behring.
Off-label drug use: Factor administration by continuous vs. bolus infusion (not product-specific discussion).
Correspondence
Claire Philipp, MD, Division of Hematology, MEB 378, UMDNJ-Robert Wood Johnson Medical School, New Brunswick, NJ 08903; Phone: (732) 235-7682; Fax: (732) 235-7115; e-mail: philipp@umdnj.edu