Abstract
Immune thrombocytopenia (ITP) comprises a heterogeneous group of disorders characterized by autoimmune-mediated platelet destruction and impairment of thrombopoiesis. ITP may occur in the absence of an evident predisposing etiology (primary ITP) or secondary to a growing list of associated conditions (secondary ITP), and must be differentiated from other causes of thrombocytopenia. This review focuses on primary ITP in adults. The traditional goal of therapy in this population is to achieve a hemostatic platelet count of 30 × 109/L or above for most patients while minimizing treatment-related morbidity. This approach has been called into question by the recent advent of well-tolerated and effective agents for the management of ITP, including pulse-dose dexamethasone, rituximab, and the thrombopoietin receptor agonists. Recent studies suggest the potential for aggressive therapy at the time of diagnosis to alter the natural history of ITP and point to the importance of quality-of-life considerations in therapeutic decision making.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune syndrome involving antibody- and cell-mediated destruction of platelets and suppression of platelet production that may predispose to bleeding.1 Recent recommendations from an international working group suggest that ITP be used to designate all cases of immune-mediated thrombocytopenia, whether occurring as a component of another clinically evident disorder or drug exposure (secondary ITP) or in the absence of a clear predisposing etiology (primary ITP).2,3 The international working group also recommends that a platelet count below 100 × 109/L, rather than 150 × 109/L, be required for diagnosis. This threshold is based on observational evidence that fewer than 10% of otherwise healthy individuals with a stable platelet count between 100 and 150 × 109/L develop more severe unexplained ITP over the ensuing 10 years.4 This review focuses on primary ITP in the adult population, but includes certain aspects of secondary forms and pediatric ITP where pertinent. The management of ITP in pregnancy is discussed elsewhere in this issue (see “Thrombocytopenia in Pregnancy”).
Incidence and Demographics
Estimates of the incidence of adult-onset ITP range from approximately 1.6 to 3.9 per 100,000 persons per year, with a prevalence ranging from 9.5 to 23.6 per 100,000 persons, based on diagnostic codes in the UK health registry5,6 ; estimates based on International Classification of Diseases, 9th revision (ICD-9) codes at hospital discharge in the United States are somewhat lower.7 However, in light of the vagaries of diagnosis and diagnostic coding, as well as the likelihood that some affected patients may not seek medical attention, the actual frequency of ITP and the number of individuals requiring therapy is uncertain.
Adult-onset ITP was once believed to be a disease largely afflicting young women. Indeed, several recent studies confirm a female preponderance in young adults, but surprisingly, they also highlight a much higher prevalence among older individuals in whom males and females appear to be equally affected.5,6 It is not known whether these findings reflect an evolution in disease epidemiology or merely a shift in the way clinicians ascertain and diagnose ITP.
Etiology
The underlying defects leading to autoantibody production are unknown. Heritability is uncommon,8 although predisposing polymorphisms in cytokines and Fcγ receptors have been described. A Th1/Th0 cytokine profile,9 a reduction in suppressor T-regulatory cells,10 and an increase in B-cell-activating factor11 may predispose to emergence of autoantibodies in response to exogenous antigens.12 Molecular mimicry appears to play a role in the development of self-reactive platelet antibodies after vaccination and certain viral infections. For example, antibodies to microbial (viral and bacterial) antigens that cross-react with platelets, often an epitope within glycoprotein IIIa, have been identified in patients who develop ITP in association with HIV, hepatitis C virus, and Helicobacter pylori infection.13–15 In support of this postulated mechanism, microbial reduction/eradication leads to remission in a substantial fraction of infected patients, although questions remain regarding how platelet-reactive autoantibodies develop and, in the case of H. pylori, why there appears to be marked variations in response rates among patients from different parts of the world.16 The mechanisms leading to autoantibody production may differ in secondary forms of ITP associated with immunosuppression or immune dysregulation.1
Pathogenesis
Platelet life span is reduced as a consequence of antibody-mediated clearance by tissue macrophages in essentially all patients. Accumulating evidence from studies of platelet kinetics also points to the contribution of immune-mediated suppression of megakaryocyte and platelet development in many patients17 ; megakaryocyte apoptosis18 and suppression of megakaryopoiesis in vitro by ITP plasma/immunoglobulin G (IgG)19 or T-cells20 ; and responsiveness to thrombopoietin receptor agonists (TRAs).21–23 Direct peroxide-induced, antibody-mediated lysis of platelets has been reported in ITP associated with HIV.24 Platelet-reactive antibodies are not detected in all individuals with ITP, and a subset of patients do not respond to pharmacologic or surgical inhibition of antibody-mediated platelet clearance or B-cell suppression, suggesting the possible involvement of other pathogenic mechanisms such as antibody-mediated apoptosis, antigen shedding, and T-cell mediated platelet destruction or marrow suppression.25
Diagnosis
Primary ITP remains a diagnosis of exclusion from both non-autoimmune causes (Table 1) and secondary causes (Figure 1) of ITP. In particular, congenital thrombocytopenias must be borne in mind in the evaluation of isolated thrombocytopenia, because differentiation from ITP can be quite difficult without historical platelet counts from the patient and study of family members. Secondary causes are estimated to comprise approximately 20% of cases of ITP in the United States, and possibly a higher percentage in regions where predisposing infections are more prevalent1 (Figure 1). Recommendations from a recent international consensus conference for the initial evaluation of primary ITP are shown in Table 2.2,3 These recommendations do not take into consideration the regional variation in response of ITP to antimicrobial therapy (e.g., the low incidence of an ITP response in the United States after eradication of H. pylori), nor the cost-effectiveness of testing in the absence of a pertinent clinical history (e.g., measuring immunoglobulin levels in all patients). Response to ITP-specific therapy remains the single most compelling diagnostic criterion.

Estimated fraction of the various forms of ITP based on clinical experience of the authors. (From Cines et al., 2009.1 Used with permission.) SLE, systemic lupus erythematosus; APS, antiphospholipid syndrome; CVID, common variable immune deficiency; CLL, chronic lymphocytic leukemia; APLS, autoimmune lymphoproliferative syndrome; post-tx, post-bone marrow or solid organ transplantation
Estimated fraction of the various forms of ITP based on clinical experience of the authors. (From Cines et al., 2009.1 Used with permission.) SLE, systemic lupus erythematosus; APS, antiphospholipid syndrome; CVID, common variable immune deficiency; CLL, chronic lymphocytic leukemia; APLS, autoimmune lymphoproliferative syndrome; post-tx, post-bone marrow or solid organ transplantation
Distinguishing primary from secondary ITP may be problematic. Patients deemed to have primary ITP on clinical grounds are often found to have antinuclear antibodies, antiphospholipid antibodies, antithyroid antibodies, or a positive direct red-cell antiglobulin test.1,26 Nevertheless, it is uncommon for patients who present with primary ITP to develop another clinically overt autoimmune disease over time, with the exception of those with antithyroid antibodies.
Natural History
There are no observational studies of untreated ITP in adults, because patients generally present with bleeding that requires intervention. The long-term outcomes in adults treated with initial daily oral prednisone supplemented with intravenous immune globulin or intravenous anti-(Rh)D are difficult to ascertain, with estimates of patients who do not require additional therapy ranging from 5% to 40%. Even patients who have required intensive therapy for years may show improvement over time.27,28 In general, however, the incidence of remission lessens as the duration of disease increases. Therefore, the international working group recommends subdividing ITP based on disease duration using the terms “newly diagnosed” for disease less than 3 months from diagnosis, “persistent” for disease lasting 3 to 12 months, and “chronic” to designate a more protracted course.2,3
Management
Principles
Treatment of ITP should be individualized. The traditional goal of therapy is to provide a hemostatic platelet count of 30 × 109/L or more for most patients, while minimizing toxicity. This approach to management is predicated on three assumptions: 1) the platelet count is a reliable surrogate marker of bleeding risk, 2) medical intervention does not alter the natural history of primary ITP, and 3) the burden of disease and its impact on quality of life are adequately captured by bleeding and drug toxicity. Each of these assumptions can be justified and each can be questioned.
Platelet Count
Evidence suggests that platelet count is predictive of serious bleeding in ITP. For example, in the absence of a hemostatic comorbidity, trauma, or surgery, intracranial hemorrhage is extremely rare in patients with platelet counts above 20 × 109/L, as is major hemorrhage in general when the platelet count exceeds 20 to 30 × 109/L. 29
On the other hand, a platelet count of 30 × 109/L may be suboptimal for some patients. Recently published recommendations from an international consensus panel state that: “Relevant factors that contribute to management decisions include the extent of bleeding, comorbidities predisposing to bleeding, complications of specific therapies, activity and lifestyle, tolerance of side effects, potential interventions that may cause bleeding, accessibility of care, patient expectations, and patient need for non-ITP medications that may create a bleeding risk.” Bleeding risk is higher in older patients and those with a prior history of bleeding.3
Remission
The potential for B- and T-epitope30 migration over time, leading to increased production of higher-affinity, platelet-specific antibodies creates a rationale for early intervention with immunosuppressive agents31 at the time of diagnosis. Recent studies using this approach appear to have challenged the concept that the natural history of ITP in adults is unalterable (see “First-line Therapy for Newly Diagnosed Patients” section below).
Quality of Life
The international consensus panel guidelines recognize the importance of patient preference.3 It is increasingly recognized that some patients with ITP experience disabling fatigue, apprehension of bleeding, restricted activities of daily living, withdrawal from important professional and recreational activities, and a poor quality of life.32,33 Two recently licensed TRAs, romiplostim and eltrombopag (see “TRAs” section below), have been shown to improve quality of life in randomized, controlled trials of adults with chronic primary ITP.21,22
Who to Treat
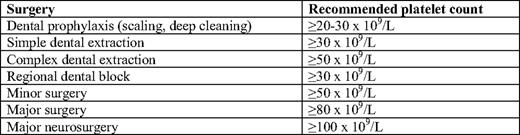
Treatment is rarely indicated at platelet counts above 30 × 109/L in the absence of platelet dysfunction or another hemostatic defect, surgery, trauma, concomitant use of anticoagulants, or in those whose lifestyle predisposes to injury.3 Consensus guidelines for target platelet counts during surgery in patients with ITP have been established by the international consensus panel (Table 3), though these are based largely on experience rather than formal study, and treatment must be individualized.
First-Line Therapy for Newly Diagnosed Patients
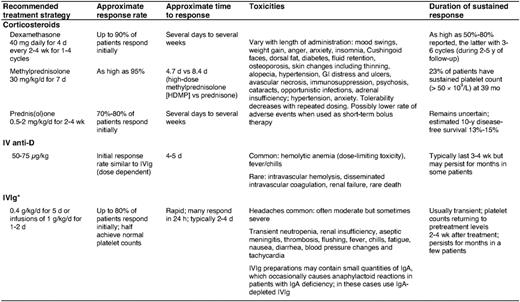
Corticosteroids, supplemented with either intravenous immune globulin or anti-Rh(D) as needed, are used to stop bleeding and increase the platelet count to above 30 to 50 × 109/L in newly diagnosed patients (Table 4). Although these agents are generally safe and well-tolerated, anti-Rh(D) administration is, on rare occasions, complicated by severe intravascular hemolysis, disseminated intravascular coagulation, and acute renal failure, and should therefore be avoided in patients with evidence of underlying hemolysis or a positive direct antiglobulin test not due to prior therapy.34 Several uncontrolled studies suggest that bolus oral dexamethasone for one to four cycles as an initial therapy increases response rates and prolongs remission duration without additional toxicity. In one trial, a single course of dexamethasone 40 mg/d for 4 d (equivalent to ∼400 mg/d prednisone) produced a sustained response in 50% of newly diagnosed adults.35 In another study, dexamethasone given every 14 d for four cycles generated an 86% response rate, with 74% of patients achieving complete remission for a median of 8 months.36 Randomized, controlled trials are needed to validate the superiority of aggressive up-front therapy over prednisone. In one recent trial, patients randomized to an intravenous anti-CD20 antibody (rituximab) and dexamethasone as an initial treatment showed higher response rates and duration than those treated with a single course of dexamethasone.31 Longer follow-up and controlled studies using optimal treatment regimes will be required to place such intense intervention at disease onset in proper perspective.
Hospitalization and Emergency Therapy
ITP patients should be hospitalized if they have: 1) internal bleeding or profound mucocutaneous bleeding, 2) platelet counts under 10 × 109/L and a history of significant bleeding or noncompliance, and 3) platelet counts of 10 to 20 × 109/L in whom responsiveness to therapy has not been established (particularly if difficulty with immediate follow-up is anticipated). Most other ITP patients can be treated as outpatients. We initiate urgent treatment with corticosteroids in combination with either intravenous immune globulin or anti-Rh(D) in Rh-positive individuals who have not undergone splenectomy and do not have a positive direct antiglobulin test, until the platelet count exceeds 30 × 109/L and bleeding, if present, stops.3,29 Platelet transfusions are often useful in the setting of life- or organ-threatening bleeding or following head trauma. We combine intravenous immune globulin and anti-Rh(D), which act through different IgG-Fcγ receptors, with vincristine and methylprednisolone in patients failing initial therapy.37 Recombinant factor VIIa is reserved for the rare patient unresponsive to other modalities if an immediate hemostatic response is necessary (e.g., intracranial hemorrhage). We use ε-aminocaproic acid or tranexamic acid to help control oral bleeding or epistaxis, topical thrombin and fibrin glue to manage dental extractions, and progestational agents to manage menorrhagia. It is also important to initiate general measures to reduce the risk of bleeding, such as cessation of drugs that impair platelet function and control of blood pressure.
Second-Line Therapy
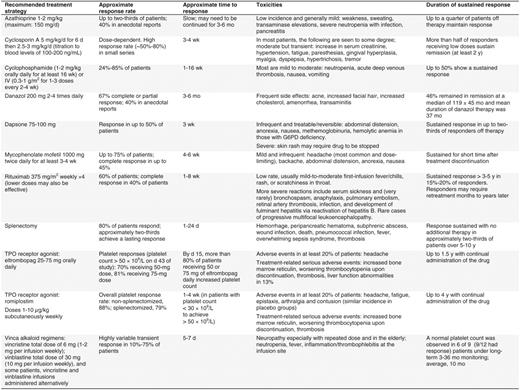
In light of the cumulative toxicities associated with prolonged corticosteroid use, minimizing steroid exposure once a response with first-line therapy occurs is a priority. Alternative modalities should be instituted in the absence of a robust response by one month or once significant steroid-related toxicity supervenes3,29 (Table 5). Occasional patients respond to extremely low levels of corticosteroids and fewer still can be treated on an every-other-day schedule. Danazol, either alone or in combination with azathioprine, and dapsone have been used as steroid-sparing agents.29 However, the next option for most patients involves splenectomy, rituximab, or TRA therapy.
Splenectomy
For decades, splenectomy has been the standard management of ITP patients unresponsive to or intolerant of prednisone. Two-thirds of patients attain a durable long-term remission, and others attain a partial response that may allow for a reduction in the use of rescue therapy.38 Even a substantial fraction of patients who do not respond to splenectomy initially may ultimately attain a hemostatic platelet count without supportive treatment.39 Morbidity and mortality of laparoscopic splenectomy are low in the hands of, experienced surgeons,40 and the incidence of overwhelming sepsis is reduced when recommended vaccination protocols are followed and antibiotics are initiated at the first sign of a systemic febrile illness.41 Response rates are lower in older patients, but there are no reliable predictors of response, with the possible exception of 111In-labeling of platelets, which is not widely available. Recently, concerns have been raised about late complications, including atherosclerosis, pulmonary hypertension, and defects in immune surveillance.38,40 Fewer physicians recommend and fewer patients now opt for splenectomy as the initial second-line form of management, although this approach by far offers the highest chance for remission and remains cost-effective.
Anti-CD20
A single course of rituximab (375 mg/m2 weekly for 4 weeks) induces a complete remission (here defined as a platelet count above 150 × 109/L) in approximately 40% of patients at 1 year,42 one-third after 2 years,43 and 15% to 20% at 5 years.3 An initial rise in platelet count often occurs within 1 to 2 weeks, suggesting an effect on platelet clearance, but more durable responses may not be observed until several months after treatment. Many patients who achieve an initial complete remission and subsequently relapse respond to retreatment. Partial responders, in contrast, often relapse within 1 year and generally do not enjoy sustained responses to retreatment. In a small, uncontrolled study, lower doses (100 mg weekly for 4 weeks) were almost as effective, although the time to response was prolonged.44 Severe side effects from infusion are uncommon, but may include respiratory distress. Rituximab is contraindicated in patients with active hepatitis B. Protracted abnormalities in B- and T-cell repertoires and impaired response to carbohydrate-based vaccine antigens have been noted, though their clinical significance is uncertain. Over 50 cases of progressive multifocal leukoencephalopathy have been reported in HIV-negative patients treated with rituximab, one of whom had ITP.45 The contribution of rituximab to the acquisition of progressive multifocal leukoencephalopathy in these heavily pretreated patients is unclear. Additional long-term safety data are required to quantify this risk and to identify risk factors for the development of this progressive and generally fatal toxicity. Other anti-CD20 antibodies are in development or clinical trials.
TRAs
Two TRAs, romiplostim and eltrombopag, have been approved by the Food and Drug Administration (FDA) for use in patients with primary ITP who require treatment after an initial course of corticosteroids. In some parts of the world, approval is restricted to patients needing therapy after splenectomy.
Romiplostim is a “peptibody” composed of four identical peptides that bind to the thrombopoietin receptor cMPL fused to an Fc fragment to prolong its half-life. It is administered weekly as a subcutaneous injection (1–10 μg/kg). In two parallel, placebo-controlled, double-blind randomized phase III trials, the study drug was given to 63 splenectomized and 62 non-splenectomized patients for 6 months. The primary efficacy end point, a platelet count of 50 × 109/L or above for at least 6 of the last 8 weeks of the study in the absence of rescue therapy, was achieved in 61% of non-splenectomized patients and in 38% of splenectomized individuals receiving romiplostim, and in only 1 of 42 subjects receiving placebo.22 Many romiplostim-treated patients reduced or discontinued concurrent ITP therapy, primarily corticosteroids, and reported improved quality of life.32 In an ongoing open-label extension study, most patients, some approaching 5.5 years of treatment with romiplostim, have sustained their platelet response.23
Eltrombopag is formulated for oral administration at a dose of 25 to 75 mg/d. It must be taken 1 h before or 2 h after a meal, and should not be taken within 4 h of medications or products containing polyvalent cations such as antacids, dairy products, or mineral supplements. Initial dosage is reduced by 50% in patients of southeast Asian origin. The dose of rosuvastatin or other substrates of the OATP1B1 transporter, which is inhibited by eltrombopag, may require adjustment. Efficacy similar to romiplostim has been observed in clinical trials of eltrombopag. In a phase III study lasting 6 weeks, 114 subjects were randomized to receive eltrombopag or placebo. The primary end point, a platelet count of 50 × 109/L or above at week 6, was achieved in 59% and 16% of eltrombopag-treated and placebo-treated subjects, respectively.21 Patients receiving eltrombopag experienced less bleeding, less need for rescue medication, and a reduction in concomitant treatments. Similar findings were documented in a 6-month phase III trial.46 Sustained responses of up to 2 years in an ongoing open-label extension study have been reported.47
Romiplostim and eltrombopag are generally well tolerated. Increased bone marrow reticulin has been observed in a few patients within 1 year of initiating TRA therapy. However, loss of response associated with evidence of a myelophthistic picture is rare and appears to be reversible in most cases. Additional long-term studies are needed to elucidate the incidence and natural history of TRA-induced bone marrow fibrosis and to compare these findings with the effects of other ITP therapies on bone marrow histology.48
Rebound thrombocytopenia to levels below those at the onset of treatment was noted in approximately 10% of patients who discontinued either romiplostim or eltrombopag in clinical trials. Gradual withdrawal of TRAs, careful surveillance, and possibly preemptive introduction of concomitant ITP medications may prevent or ameliorate this toxicity.
To date, there is no compelling evidence that either TRA increases thromboembolic complications in patients with ITP. In a pooled analysis of all studies of romiplostim, the incidence of thrombosis did not differ among those treated with study drug and those receiving placebo (8 vs. 10 events/100 patient-years, respectively).49 In a pooled analysis of studies of eltrombopag in ITP, 17 eltrombopag-treated patients suffered thrombotic complications over 377 patient-years of exposure. Although no events were documented in placebo-treated subjects, the exposure of this cohort was limited to only 26 patient-years.50 Most TRA-associated thromboembolic events have occurred in patients with preexisting atherosclerosis or thrombotic risk factors, often in the setting of a low or normal platelet count. In light of recent evidence that eltrombopag may increase the risk of venous thrombosis in patients with hepatitis C, and that ITP itself may be prothrombotic in some patients,51 additional study of the incidence of TRA-associated thrombosis and identification of potential risk factors is warranted.
Thus far, data regarding the risk of leukemogenesis with TRA therapy are reassuring. In the controlled trials of romiplostim and eltrombopag, the incidence of hematologic neoplasm was low and similar in the treatment and placebo groups.21,22,46 In studies of patients with myelodysplastic syndrome-associated thrombocytopenia, romiplostim was associated with a transient and reversible rise in circulating blasts in a minority of patients, but the rate of leukemic transformation did not differ between romiplostim- and placebo-treated subjects.52
Hepatobiliary laboratory abnormalities were detected in 13% of eltrombopag-treated patients, prompting a black-box warning that calls for regular monitoring of liver function tests. Cataract formation was observed in preclinical testing of eltrombopag in juvenile rodents at doses several times higher than the human clinical exposure. Until more is known regarding the risk of cataract development in humans at clinically approved doses of eltrombopag, periodic ophthalmic examinations are advisable.
Third-Line Approaches
Several immunosuppressive agents alone or in combination have shown some efficacy in small, uncontrolled studies; these include azathioprine, cyclophosphamide, mycophenylate mofetil, and cyclosporine3,53 (Table 5). However, because of a less favorable safety profile, these agents are generally reserved for patients unresponsive to or ineligible for the aforementioned first- and second-line treatment options. Vinca alkaloids are used infrequently and hematopoietic stem-cell transplantation is reserved for extreme circumstances. New agents that affect antibody production, platelet production, or clearance are in development.
Summary and Future Directions
ITP is a syndrome of various disorders that have in common immune-mediated thrombocytopenia, but that differ in pathogenesis, natural history, comorbidities, and responsiveness to therapy. As our understanding of disease pathogenesis grows, the term “primary ITP” will be confined to a decreasing proportion of patients in whom a potential etiology has not been identified and pathogenic pathways have not been elucidated.1 The contribution of impaired platelet poiesis remains uncertain and cannot as yet be monitored clinically, but TRAs appear capable of overriding antibody-mediated platelet clearance in most patients. It may be possible to use high-throughput screening to clone autoantibodies and identify epitopes to search for external antigens that predispose to platelet antibody formation for the purposes of developing rational therapies. Identification of the responsible B- and T-cell clones would also enhance drug monitoring and might lead to more selective therapy.
Approaches to the management of ITP remain in flux. In the absence of predictors of bleeding or biomarkers of pathogenesis or response, platelet counts remain the surrogate marker of choice, however imprecise. The international working group report notes that in patients with persistent ITP “…spontaneous remission may still occur and aggressive interventions might be deferred, if possible.” This recommendation is being challenged by the high complete remission rates observed in studies of up-front aggressive therapy, although the long-term benefit and toxicity of such approaches remain to be determined through controlled studies with long-term follow-up.
With regard to second-line therapy, a minimalist approach based on bleeding and a threshold platelet count of 30 × 109/L using prednisone followed by splenectomy is cost-effective and has been proven to be clinically effective in terms of preventing death from hemorrhage. However, the toxicity of corticosteroids and concerns about the long-term effects of splenectomy have affected patient and physician preferences and are associated with impaired quality of life in non-responders. Rituximab has a favorable toxicity profile, but the effect of repetitive cycles of immunosuppressive therapy has not been assessed in large numbers of patients or with follow-up comparable to splenectomy. TRAs pose an exciting new treatment option and a new set of challenges with respect to duration of treatment, cost, and need for long-term safety studies. Newer agents that affect antibody production, platelet production, and clearance are in development. The last few years have been marked by tremendous advances in our understanding and management of ITP; fortunately, it appears that this is likely to continue.
Disclosures
Conflict-of-interest disclosure: DBC has consulted for Glaxo-Smith-Kline and Amgen. Off-label drug use: Only intravenous immune globulin G, anti-Rh(D), romiplostim, and eltromopag are approved for the treatment of ITP. All other agents for the management of ITP discussed in this chapter are off-label.
Correspondence
Adam Cuker, MD, MS, Hospital of the University of Pennsylvania, 3 Dulles, 3400 Spruce Street, Philadelphia, PA 19104; Phone: (215) 615-8015; Fax: (215) 615-6599; e-mail: adam.cuker@uphs.upenn.edu