Abstract
Thrombocytopenia occurs commonly during pregnancy, and may result from diverse etiologies. Awareness of these many causes facilitates proper diagnosis and management of thrombocytopenia in the pregnant setting. Some causes of thrombocytopenia are unique to pregnancy and may not be familiar to hematologists. In the review, we will discuss the differential diagnosis of thrombocytopenia in pregnancy, and the pathogenesis of selected thrombocytopenic disorders. Considerations for optimal management of the pregnant patient with thrombocytopenia will also be described.
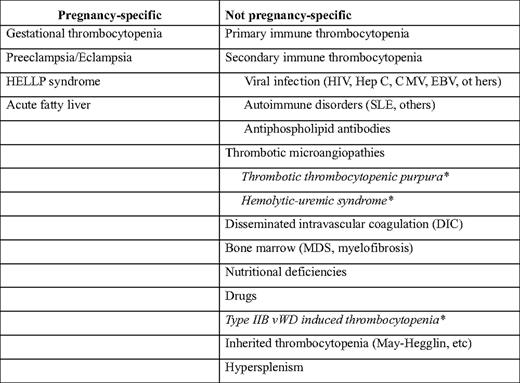
Thrombocytopenia affects 6% to 10% of all pregnant women and other than anemia is the most common hematologic disorder in pregnancy. There are many potential causes of pregnancy-associated thrombocytopenia (Table 1); some of these are unique to pregnancy, whereas others may also occur in the nonpregnant setting.1 Pregnancy is associated with numerous metabolic, immunologic, and other homeostatic changes that require careful consideration when attempting to define the cause of thrombocytopenia in a particular individual. Moreover, because therapeutic interventions used to treat thrombocytopenic disorders in pregnant women may have toxicities unique to pregnancy, management approaches must be carefully considered.
We will review the differential diagnosis of thrombocytopenia in pregnancy, discuss the relevant pathophysiologic mechanisms, and suggest strategies for optimal management. Because a complete discussion of the many causes of thrombocytopenia in pregnant women is beyond the scope of this review, we will provide an overview of the more common disorders, emphasizing disorders such as preeclampsia, in which recent advances have provided new insight into underlying mechanisms.
Disorders That Cause Thrombocytopenia in Pregnancy
Gestational Thrombocytopenia
Gestational thrombocytopenia, also known as incidental thrombocytopenia of pregnancy, is the most common cause of thrombocytopenia in pregnant women, accounting for approximately 75% of all cases.1
Normal pregnancy is associated with a physiologic fall in the platelet count that is characterized by a leftward shift in the platelet count distribution (Figure 1).2 The reason for this decline is unknown, although it has been speculated that these changes may reflect dilution, decreased platelet production, or increased platelet turnover during pregnancy.3 Regardless, the fall in the platelet count during normal pregnancy results in some pregnant women developing platelet counts that fall into the thrombocytopenic range.2 Generally, these individuals have mild thrombocytopenia that first becomes apparent in the mid-second to third trimester of pregnancy. Although there is no well-established minimum value for the platelet count in gestational thrombocytopenia, most experts consider this diagnosis to be less likely when the platelet count falls below 70,000/μL. However, reports exist of more severe thrombocytopenia in pregnant women that was not responsive to steroid therapy and resolved postpartum, and thus was consistent with gestational thrombocytopenia.

Histogram of platelet counts from pregnant (open bars) and nonpregnant women (filled bars). Reproduced from Boehlen et al,2 with permission.
Histogram of platelet counts from pregnant (open bars) and nonpregnant women (filled bars). Reproduced from Boehlen et al,2 with permission.
Because there is no diagnostic testing available for gestational thrombocytopenia, this disorder is a diagnosis of exclusion.4 Patients with a history of primary or secondary immune thrombocytopenia (ITP), thrombocytopenia of any etiology preceding pregnancy, or any reason for thrombocytopenia other than uncomplicated pregnancy itself are generally not considered to have gestational thrombocytopenia. However, in many cases, it may not be possible to distinguish gestational thrombocytopenia, particularly a more severe case, from ITP.
Gestational thrombocytopenia is not associated with adverse outcomes to either the mother or fetus.5 The incidence of fetal or neonatal thrombocytopenia in the offspring of such patients is no higher than that of nonthrombocytopenic women, and when it occurs often results from coincident neonatal alloimmune thrombocytopenia.4,5 The degree of maternal thrombocytopenia is generally not severe enough to increase the risk of bleeding with delivery, although some cases may compromise the ability to deliver epidural anesthesia. Because such cases may be difficult to distinguish from ITP, a short trial of ITP therapy (eg, corticosteroids or intravenous immunoglobulin [IVIg]) may be useful both diagnostically and therapeutically. In the absence of a platelet increment, platelet transfusion may be used to raise the platelet count to a level deemed safe for epidural catheter placement if desired.3 Gestational thrombocytopenia is self-limited and resolves within 1 to 2 months after delivery.
Immune Thrombocytopenia
ITP is an uncommon cause of thrombocytopenia in pregnancy, occurring in between 1 in 1000 and 1 in 10,000 pregnant women.6 As opposed to secondary ITP, which develops in association with viral infection (human immunodeficiency virus [HIV], hepatitis C, and Helicobacter pylori), autoimmune disease, and other challenges, factors that induce primary ITP and why its course worsens in some pregnant patients are not well understood. Of all cases of pregnancy-associated ITP, approximately one-third is first diagnosed during pregnancy, whereas two-thirds are in patients with preexisting disease.
The clinical features of ITP in a pregnant patient are similar to those encountered in nonpregnant women, with bruising, mucosal bleeding, and petechiae as presenting symptoms whose severity parallel the degree of thrombocytopenia. Although ITP may present at any point in pregnancy, it is one of the few causes of thrombocytopenia that may become manifest in the first trimester.7 Because ITP may be indistinguishable from gestational thrombocytopenia, patients with ITP in pregnancy often have a prior history of this or other immune-mediated disorders.5,7,8
The goal of therapy for ITP in pregnant women is to prevent bleeding. Thus, treatment is generally not required in patients with platelet counts greater than 20,000 to 30,000/μL who are not bleeding. In one large series of pregnant women with ITP, only approximately 30% required therapy.9 However, patients who wish to receive epidural anesthesia, which may increase the risk of epidural hematoma formation, require higher platelet counts and a more aggressive approach. Although no evidence-based recommendations exist, recent guidelines recommend a platelet count of at least 75,000/μL for safe placement of an epidural catheter.3 Some advocate maintaining a platelet count of at least 50,000/μL beyond the mid- to late third trimester in the event that an unplanned cesarean section may be required.
Corticosteroids, the first-line of therapy for ITP in nonpregnant individuals, are equally efficacious in pregnant women, with response rates of 70% to 80%. However, corticosteroids cause several unique toxicities in pregnancy, such as gestational diabetes and pregnancy-induced hypertension. These agents may also be associated with premature rupture of the fetal membranes and placental abruption,10 and some reports implicate exposure to high doses of corticosteroids in the first trimester with developmental anomalies, such as orofacial clefts. For these reasons, corticosteroids should be used sparingly in pregnancy, with the minimal effective doses employed. Others have cogently argued that, due to the toxicity of corticosteroids, IVIg should be considered the first-line of therapy for pregnancy-associated ITP.6 In general, the advantages and disadvantages of each agent must be carefully considered in the context of factors such as when within the gestational period therapy is needed (ie, during the first trimester), the expected duration of therapy, and specific characteristics of the patient (ie, risk factors for pregnancy-induced hypertension). Intravenous anti-D has also been used effectively for therapy of ITP in pregnancy, although in only a small number of reported cases.11
For patients who do not respond to corticosteroids or IVIg as single agents, combinations of these therapies may sometimes be more effective, particularly when corticosteroids are delivered as high-dose “pulse” therapy (eg, methylprednisolone, 1 g/day for two consecutive days).3 If this approach fails, laparoscopic splenectomy may be safely performed during pregnancy. If splenectomy is needed, it should be performed during the mid-second trimester, if possible, to avoid early pregnancy loss and obstruction of the surgical field by the gravid uterus at later dates.
For pregnant patients with refractory ITP, toxicity and teratogenicity of therapeutic agents limits available options. Azathioprine has been used safely during pregnancy in patients with renal transplants and inflammatory bowel disease. Cyclosporin A has also been used safely in pregnancy. Most cytotoxic agents are teratogenic, but have been used in patients with neoplastic disease during the mid-second trimester and beyond. The thrombopoietic agents, romiplostim and eltrombopag, are considered category C in pregnancy, with little to no data available concerning their safety. Rituximab has been used for ITP in pregnancy, but caused a delay in neonatal B-lymphocyte maturation, although without apparent clinical consequences.12
Antiplatelet antibodies cross the placenta and can induce thrombocytopenia in the fetus. The development of fetal thrombocytopenia is dependent upon many poorly understood factors, such as the maturity of the fetal reticuloendothelial system, and cannot be predicted using available clinical or laboratory parameters. There is no consistent correlation between neonatal thrombocytopenia and the severity of maternal thrombocytopenia, the level of maternal platelet-associated immunoglobulin, whether or not the mother has undergone splenectomy, or several other parameters.7,8,10 In fact, the best predictor of neonatal thrombocytopenia identified to date is a history of thrombocytopenia in a prior sibling.13
Thrombocytopenia, with a platelet count < 100,000/μL, develops in approximately 15% of the offspring of mothers with ITP. Ten percent of neonates develop more severe thrombocytopenia, with platelet counts below 50,000/μL, and platelet counts below 20,000/μL occur in approximately 4%.8 Neonates with severe thrombocytopenia may experience bleeding complications, the most feared of which is intracranial hemorrhage, particularly as a consequence of head trauma during vaginal delivery. In previous decades, this concern dictated the delivery approach, with all patients undergoing cesarean section.3 With the advent of techniques for accurate measurement of the fetal platelet count by percutaneous umbilical cord blood sampling (or cordocentesis), this approach evolved into one in which vaginal delivery was allowed in the absence of significant fetal thrombocytopenia. However, the results of several studies demonstrated that (i) the risk of fetal intracranial hemorrhage in the offspring of mothers with ITP is very low—between 0% and 1.5%8 ; (ii) there is no evidence this risk is increased by vaginal delivery,14 and (iii) the risk of complications that may lead to urgent cesarean section and compromised fetal outcomes with cordocentesis (1%–2%) is at least as high as the risk of fetal intracranial hemorrhage due to maternal ITP.7 These observations have been incorporated into current guidelines, which suggest that the mode of delivery in pregnant patients with ITP should be dictated solely by maternal indications,3 and the fetal platelet count not be routinely determined. However, the neonatal platelet count should be determined in cord blood immediately on delivery for the next 5 days, because the nadir neonatal platelet count may not develop until several days after birth. Neonates with platelet counts < 50,000/μL, even if asymptomatic, should undergo transcranial ultrasound to exclude intracranial hemorrhage.3
Preeclampsia and the HELLP Syndrome
These disorders are discussed together, because they share similar clinical characteristics and pathophysiology. Preeclampsia, defined by criteria established by the American College of Obstetricians and Gynecologists, affects up to 6% of all first pregnancies15 and is the most common cause of pregnancy-associated mortality worldwide. Criteria for the diagnosis of preeclampsia include the following: (i) blood pressure of 140 mm Hg systolic or 90 mm Hg diastolic or higher that occurs after 20 weeks of gestation in a woman with previously normal blood pressure; and (ii) proteinuria, defined as urinary excretion of 0.3 grams of protein or higher in a 24-hour specimen, that usually correlates with a 1+ or greater reading on dipstick.15 Multiple organ systems are affected in preeclampsia, reflecting systemic endothelial dysfunction, although the kidneys are affected most severely.16 Predisposing factors include age < 20 years or > 30 years, a high body mass index, chronic hypertension, and a history of insulin resistance, among others. Both maternal and paternal genetic factors also contribute.
Thrombocytopenia occurs in up to 50% of women with preeclampsia, and its severity generally parallels that of the underlying preeclampsia. Importantly, thrombocytopenia may occasionally precede other manifestations of preeclampsia, and thus preeclampsia must be considered in the differential diagnosis of isolated thrombocytopenia developing in the late second or third trimester.
In recent years, several studies have provided insight into the pathogenesis of preeclampsia. Although the clinical manifestations of preeclampsia usually do not appear until the late second or third trimester, the pathologic processes underlying this syndrome reflect inadequate placentation early in pregnancy. Placentation, the process by which fetal trophoblast cells invade the maternal decidua and remodel the maternal uterine spiral arteries, is necessary to ensure an adequate placental blood supply. In preeclampsia, both the depth of trophoblast invasion and the extent of remodeling of the spiral arteries are reduced (Figure 2); this may reflect deficiencies in trophoblast function, including failure of trophoblast cells to alter their pattern of integrin expression toward an endothelial phenotype and deficient protease activity, among others.17 Insufficient placentation results in progressive ischemia of the fetoplacental unit as pregnancy advances, and fetal growth and oxygen requirements increase.

Abnormal placentation in preeclampsia. Note the decreased trophoblastic invasion at decidual and myometrial levels, narrower vascular lumen of the maternal uterine spiral artery, and failure of trophoblast cells to replace the maternal endothelium lining of the spiral artery in the preeclamptic placenta. Reproduced from Kita and Mitsushita,20 with permission.
Abnormal placentation in preeclampsia. Note the decreased trophoblastic invasion at decidual and myometrial levels, narrower vascular lumen of the maternal uterine spiral artery, and failure of trophoblast cells to replace the maternal endothelium lining of the spiral artery in the preeclamptic placenta. Reproduced from Kita and Mitsushita,20 with permission.
Although deficient trophoblast invasion as a critical component of preeclampsia was recognized many years ago, the mechanisms by which uteroplacental hypoxia induce the systemic manifestations of preeclampsia have only more recently begun to be unraveled. Recent studies have demonstrated that increased plasma levels of soluble vascular endothelial cell growth factor (VEGF) receptor type 1 (sFlt1),18 as well as endoglin, an endothelial cell-derived member of the tumor growth factor-β (TGF-β) receptor family,19 are present in patients destined to develop preeclampsia as early as the late first trimester. Increased levels of sFlt1 and endoglin mRNA are present in preeclamptic placentae, suggesting this is the source of these proteins.18,20 sFlt1 binds and neutralizes VEGF and placental growth factor (PLGF), another important VEGF family member whose levels normally increase during pregnancy, whereas endoglin blocks the binding of TGF-β to endothelial cells.17 One outcome of these actions is to decrease expression of endothelial nitric oxide (NO) synthase leading to reduced NO production and exacerbation of the hypertensive manifestations of preeclampsia. Functional deficiency of VEGF/PLGF also results in endothelial dysfunction, particularly that of the glomerular endothelium, leading to the characteristic endothelial swelling and “glomerular endotheliosis” lesions of preeclampsia and the development of a thrombotic microangiopathy.21
The syndrome of Hemolysis, Elevated Liver function tests, and Low Platelets (HELLP) affects 0.5% to 0.9% of all pregnancies and develops in 10% of patients with preeclampsia.22 Although the specific pathophysiology of HELLP has not been as closely studied as that of preeclampsia, the disorders share clinical features, and likely, pathogenesis.23 Criteria for the HELLP syndrome vary somewhat among studies, but generally include microangiopathic hemolytic anemia, a lactate dehydrogenase (LDH) > 600 U/mL, increased aspartate aminotransferase (≥ 40–70 U/mL, depending on the series), and thrombocytopenia (platelet count < 100,000/μL, or in some series, 150,000/μL).24 Unlike preeclampsia, HELLP is more common in multiparous women. Approximately 70% of cases of HELLP occur prior to term, with the remainder usually occurring within 48 hours after delivery.22 Up to 75% of patients with HELLP have proteinuria, but only 50% to 60% have hypertension. Patients with HELLP frequently present with malaise, accompanied by severe right upper quadrant pain thought to result from obstruction of blood flow in the hepatic sinusoids24 ; liver pathology confirms hepatocyte necrosis with fibrin deposition in periportal sinusoids.25 A wide differential diagnosis must be considered in patients presenting with HELLP, including intraabdominal processes such as nephrolithiasis, cholecystitis, and appendicitis, among others.22 A recent study identified mutations in genes regulating the activity of the alternative complement system (factor H, factor I, and membrane cofactor protein) in 4 of 11 patients with HELLP,26 suggesting that, as in atypical hemolytic uremic syndrome (HUS), excessive complement activation may be involved in the pathogenesis of HELLP in some patients (vide infra).
For detailed discussion of the management of preeclampsia and HELLP, readers are referred to several excellent reviews.22,27 The general approach to therapy for these disorders in women beyond 34 weeks is medical stabilization, followed by expeditious delivery of the fetus. At earlier points in pregnancy, delivery may be undertaken following administration of betamethasone to enhance fetal lung maturity, although some advocate expectant therapy in selected patients as a means to improve fetal outcome. Both disorders generally begin to remit within several days after delivery, although in some individuals, in particular those with HELLP, prolonged thrombocytopenia and elevations of LDH lasting for as long as several weeks may occur postpartum.25 HELLP and preeclampsia may also uncommonly present postpartum. The use of plasma exchange and corticosteroids in such patients with persistent postpartum thrombotic microangiopathic syndromes has been demonstrated in small studies to induce a more rapid remission, although little controlled data are available.
A patient with preeclampsia or HELLP is at increased risk for development of recurrent disease and poor pregnancy outcome in subsequent pregnancies, particularly if the initial disorder occurred with an early onset.28 Several large clinical trials have utilized agents such as aspirin or antioxidants such as vitamins C and E, in attempts to prevent preeclampsia, particularly in high-risk individuals. A meta-analysis suggests that aspirin has modest efficacy in prevention of preeclampsia, although no difference in the incidence of fetal death was demonstrated.29 Antioxidant trials have also been disappointing. In recent years, it has become increasingly apparent that patients with preeclampsia are at increased risk for cardiovascular disease in long-term follow-up.
Thrombotic Thrombocytopenic Purpura and HUS
Thrombotic thrombocytopenic purpura (TTP) and HUS, collectively referred to as thrombotic microangiopathies (TMAs), are not pregnancy-specific, although they occur with increased frequency during or in relation to pregnancy.30 In many large series, 20% or more of the included patients developed disease during pregnancy or the immediate postpartum period. TTP and HUS share many overlapping features and may be difficult to discern from one another. Microangiopathic hemolytic anemia and thrombocytopenia are also common to pregnancy-specific disorders such as preeclampsia, the HELLP syndrome, and acute fatty liver. Thus, the differential diagnosis in a pregnant patient presenting with such manifestations is complex, and, in some cases, achieving a definitive diagnosis may not be possible.31,32
TTP is classically characterized by the pentad of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, neurologic dysfunction, fever, and renal disease. However, due to the importance of initiating therapy promptly for this disorder, any patient with MAHA and thrombocytopenia that is otherwise unexplained should be considered to have TTP. Of the other clinical manifestations, neurologic dysfunction is most common in classical TTP.33 Involvement of organs such as the pancreas, heart, and others are demonstrable pathologically, and may lead to complications such as pancreatitis, myocardial infarction, and cardiac arrythmias. TTP is strongly associated with a severe deficiency of ADAMTS-13, a metalloprotease that cleaves ultra-large von Willebrand factor (VWF) multimers, the most hemostatically active species of VWF. Deficiencies of ADAMTS-13 are usually acquired, resulting from neutralizing autoantibodies,34 although congenital ADAMTS-13 deficiency (Upshaw-Schulman syndrome) accounts for a minority of cases.35 Increased levels of ultra-large VWF species present in the setting of ADAMTS-13 deficiency promote platelet agglutination and thrombotic occlusion of the microvasculature.
Why the incidence of TTP is increased in pregnancy is uncertain. Whereas a physiologic decrease in ADAMTS-13 levels occurs in pregnant women, these remain well above the levels of 5% to 10% that are associated most strongly with TTP. Some studies suggest that the greatest incidence of TTP occurs during the mid-second trimester,36 although others report a higher incidence in the third trimester of pregnancy. Patients with pregnancy-associated TTP are at increased risk for the development of recurrent TTP in subsequent pregnancies.
The management of TTP during pregnancy is much like that in the nonpregnant patient, with plasma exchange yielding a response rate of approximately 80%. The role of corticosteroids in the management of TTP has not been determined through randomized studies; although the immune nature of the disorder provides a rationale for their use, corticosteroids are associated with an increased risk of complications in pregnant individuals (see section on ITP). TTP occurs in a high proportion of patients with Upshaw-Schulman syndrome and is associated with high fetal mortality; periodic plasma infusions appear to be helpful, but a specific protocol has not been developed and treatment must remain empiric (ie, aimed at maintaining ADAMTS13 levels above 15%).37 Importantly, unlike preeclampsia and the HELLP syndrome, termination of pregnancy does not induce remission of TTP.38
HUS encompasses the same pentad of symptoms as TTP, although as with TTP, all of the symptoms are usually not present. Although a number of classification schemes have been proposed for the TMAs, the variant of HUS that occurs most commonly in association with pregnancy is consistent with “atypical,” diarrhea negative (D-) HUS. Most patients with HUS have a more prominent component of renal insufficiency than those with TTP, although significant overlap in symptoms/findings may occur. Levels of ADAMTS-13 are generally not severely reduced in most patients with HUS, although the same is also true of some patients with clinical features most consistent with classic TTP. Thus, distinguishing the two disorders based on presenting clinical features alone may be difficult.31,32 One feature that may be useful in distinguishing these disorders in the setting of pregnancy is that, unlike TTP, most cases of HUS develop several weeks postpartum.
Atypical HUS has been associated with abnormalities in regulation of the alternative pathway of complement. Underlying this association has been the identification of mutations in complement regulatory proteins such as factor H, factor I, membrane cofactor protein, and thrombomodulin, as well as activating mutations in complement factors B and C3. Acquired deficiencies of factor H have also resulted from factor H-reactive antibodies.39 In a retrospective report, the significance of altered complement regulation in pregnancy-associated HUS was evaluated.40 Key findings were that (i) of 100 adult female patients with atypical HUS, 21% had experienced pregnancy-associated atypical HUS; (ii) consistent with previous reports, 79% of atypical HUS cases occurred during the postpartum period; (iii) complement abnormalities were detected in 18 of the 21 patients with pregnancy-associated atypical HUS; (iv) the risk of atypical HUS was greatest during the second pregnancy; and (v) overall outcomes of atypical HUS, which included 76% of patients developing end-stage renal disease, were not affected by whether the patient was pregnant at the time of diagnosis. Of 44 pregnancies in patients with complement abnormalities, 4.8% and 7.7% were associated with fetal loss or preeclampsia, respectively.40 The poor outcomes in this series demonstrate the gravity of this syndrome, which usually responds poorly to plasma exchange and leaves a high proportion of patients with chronic renal insufficiency. Nevertheless, a clinical trial of plasma exchange is reasonable, particularly in light of the difficulties in distinguishing atypical HUS from TTP. Other interventions, including anticoagulants and antiplatelet therapies, have not been beneficial. The outcomes of studies that examine the utility of inhibitors of complement activation are awaited with interest. Testing for inherited abnormalities of complement regulation in women with family members affected by atypical HUS during pregnancy should be considered.
Miscellaneous Causes of Thrombocytopenia
In this section, we will briefly discuss several miscellaneous causes of thrombocytopenia in pregnancy. These, and other causes that are not discussed, are listed in Table 1.
In all cases of thrombocytopenia, whether in pregnant or nonpregnant individuals, the peripheral blood film should be examined closely to evaluate for ethylenediaminetetraacetic acid-dependent platelet clumping, causing “pseudothrombocytopenia.” In such cases, determination of the platelet count in a citrate tube may eliminate clumping and lead to more accurate readings.
Disseminated intravascular coagulation (DIC) may arise from a number of events in pregnant women. Some causes of fulminant DIC include placental abruption, amniotic fluid embolism, and uterine rupture; in each of these situations, a rapid release of tissue factor-rich material into the maternal circulation leads to profound activation of coagulation, with consumption of coagulation factors and severe hypofibrinogemia. DIC may also be present in association with retained fetal products, but in these cases is often compensated and more gradual in onset, and thrombocytopenia may be the presenting symptom.25 An increased D-dimer level may be useful diagnostically.
Acute fatty liver of pregnancy is a rare disorder that usually presents in the third trimester of pregnancy with nausea, vomiting, malaise, right upper quadrant pain, and cholestatic liver dysfunction.41 Most patients develop DIC due to acquired antithrombin deficiency, with thrombocytopenia and deficiencies of fibrinogen and other clotting factors. Due to the coagulopathy, bleeding is common, despite only mild thrombocytopenia. Some cases of acute fatty liver, as well as HELLP, may be associated with fetal mitochondrial fatty acid oxidation defects, most commonly due to deficiency of long-chain 3-hydroxyacyl coenzyme A dehydrogenase. Treatment involves supportive care with blood product support for the underlying coagulopathy.1
Type IIb von Willebrand disease is characterized by a mutant VWF molecule that binds to its primary platelet receptor, glycoprotein Ib, with increased affinity, thereby inducing platelet agglutination, accelerated platelet clearance, and thrombocytopenia. A number of underlying mutations, all in exon 28, have been described (http://www.vwf.group.shef.ac.uk). During pregnancy, levels of endogenous mutant VWF increase in response to the estrogen-rich hormonal milieu, and thrombocytopenia induced by the mutant VWF may become more apparent. In occasional patients, platelet counts have been reported to fall to as low as 20,000 to 30,000/μL, with improvement after delivery.
In addition to primary ITP, ITP secondary to infection (HIV, hepatitis C, cytomegalovirus, H pylori, Epstein-Barr virus, etc), or autoimmune disorders—such as systemic lupus erythematosus or the antiphospholipid syndrome—should also be considered in the differential diagnosis.1 Antiphospholipid antibodies have been associated not only with a secondary ITP, but also a microangiopathic thrombocytopenia as well.42 Drug-induced thrombocytopenia may result from a variety of prescription drugs, as well as illicit medications, such as cocaine. Myelopthistic causes of thrombocytopenia, although unlikely in women of child-bearing age, include infiltrative marrow disorders (eg, metastatic neoplasms, fungal, or other types of infections), as well as primary bone marrow syndromes (including myelofibrosis, myelodysplasia, and related disorders). Inherited thrombocytopenia, such as the May-Hegglin anomaly, may first be detected during pregnancy, and may be detected through identification of abnormal platelet morphology on review and assessment of the peripheral blood film of family members.1
Disclosures
Conflict-of-interest disclosure: The author is associated with Speaker Bureaus for Amgen and GlaxoSmithKline, and is a member of GlaxoSmithKline's advisory committees.
Off-label drug use: None disclosed.
Supplied by Grants HL072033
Correspondence
Keith R. McCrae, MD, Professor, Hematologic Oncology & Blood Disorders, The Cleveland Clinic Foundation, 9500 Euclid Ave., NC10, Cleveland, OH 44195; Phone: (216) 445-7809; Fax: (216) 444-9404; e-mail: mccraek@ccf.org