Abstract
Since the introduction of replacement coagulation factor infusions for the treatment of hemophilia in the 1970s and subsequent improvements in the safety profile of available factor VIII (FVIII) and factor IX (FIX) concentrates, mortality among patients with hemophilia has improved considerably and now parallels that of the noncoagulopathic population in developed countries. Substantial morbidity, however, continues from the development of inhibitory antibodies, a recognized complication of clotting factor replacement; from infections and thrombosis complicating placement of central venous catheters, which are required in children with hemophilia due to frequent prophylactic infusions of coagulation factors with defined half-lives; and from disabling joint disease in individuals without access to costly prophylaxis regimens. In response to the need for long-acting, more potent, less immunogenic, and more easily administered therapies, an impressive array of novel agents is nearly ready for use in the clinical setting. These therapeutics derive from rational bioengineering of recombinant coagulation factors or from the discovery of nonpeptide molecules that have the potential to support hemostasis through alternative pathways. The number of novel agents in clinical trials is increasing, and many of the initial results are promising. In addition to advancing treatment of bleeding episodes or enabling adherence to prophylactic infusions of clotting factor concentrate, newer therapeutics may also lead to improvements in joint health, quality of life, and tolerability of iatrogenic or comorbidity-associated bleeding challenges.
Introduction
In the developed world, aggressive regimens of early administration of prophylactic factor concentrate have led to dramatic advances in joint health and quality of life among boys with hemophilia.1–3 An equally encouraging improvement in the average life expectancy of adult males with hemophilia, now approaching that of the noncoagulopathic population, is characteristic of the modern era, thanks to the widespread availability of replacement clotting factor concentrate and effective methods of eradicating or controlling infection with hepatitis C virus and HIV.4 Now that many of the devastating effects of the heritable bleeding disorder and, in some case, its treatment, can be mitigated, leading to reduced morbidity and mortality among pediatric and adult patients who are treated with optimal protocols, further improvements in the medical therapy of congenital deficiency of coagulation factor VIII (FVIII) or IX (FIX) can be explored. The focus of this review is the development of novel therapeutic agents, including modified coagulation proteins and nonpeptide hemostatic agents, that may improve the prevention or treatment of hemophilia-related bleeding episodes; further reduce hemophilia-related morbidity, including hemophilic arthropathy; and continue to improve health-related quality of life through the availability of more effective, better tolerated, or safer hemostatic agents.
Bioengineering of therapeutic coagulation proteins
The impetus for biochemical modification of existing therapeutic hemostatic proteins (ie, coagulation factors) derives from clinical observations of the limitations of the currently available therapies. Despite advances that have led to the availability of purified plasma-derived and recombinant FVIII5 and FIX,6 infusions of replacement coagulation factors require intravenous access, must be given repeatedly and frequently due to defined terminal half-lives of the replacement protein, and are associated with the development of inhibitory antibodies in 25%-30% of patients with hemophilia A7 and to a lesser extent in patients with hemophilia B.6 Patients who develop inhibitory antibodies to FVIII or FIX that do not respond to immune tolerance induction incur severe morbidity due to repeated episodes of joint and soft tissue bleeding.8 For those who select rFVIIa as treatment for bleeding episodes, normalization of hemostasis is constrained by the short half-life of the molecule.9 Furthermore, the initial efficacy of either rFVIIa or the currently available activated prothrombin complex concentrate for the treatment of bleeding events is variable, leading to poor responses in some patients.10
Efforts to rationally modify currently available replacement clotting factor concentrates reflect a desire to increase half-life, improve ease of delivery, reduce immunogenicity, and/or increase potency11 (Table 1). Contemporary strategies to improve coagulation factor therapeutics in hemophilia include chemical modification, fusion to biologic proteins, and site-directed mutagenesis (Table 2).
Chemical modification
PEGylation
PEGylation involves the covalent conjugation of polyethylene glycol (PEG) to a therapeutic protein. The PEG polymers create a diffusion cloud around the protein, shielding it from exposure to proteolytic enzymes, clearance receptors, and immune effector cells (which otherwise might recognize antigenic peptide epitopes).32 The strategy has been effective at prolonging the circulating half-life of a substantial number of approved, widely used biopharmaceuticals, allowing for less frequent administration.32 However, careful design of the PEGylated protein is required because the PEG polymers may interfere with the peptide's interaction with substrates, reducing the biologic activity of the protein.33 This phenomenon was observed in a recombinant B domain–deleted rFVIII (BDD-rFVIII) to which PEG polymers were randomly conjugated.34 The PEGylated BDD-rFVIII exhibited reduced binding to VWF, which, paradoxically, could lead to a shortened half-life.35 This limitation was addressed by introducing surface-expressed cysteine residues into BDD-rFVIII, providing substrates for site-directed PEGylation.36 Screening of the resulting targeted PEGylated variants revealed some with full in vitro coagulant activity and VWF binding, and in hemophilia A mice and rabbits, certain PEGylated FVIII muteins showed almost doubling of the terminal half-life compared with non-PEGylated BDD-rFVIII. In addition, improved survival following tail transaction in hemophilia A mice that had received PEGylated BDD-rFVIII compared with those who had received standard rFVIII was observed.36 One such BDD-rFVIII molecule, modified by conjugation to a single, branched PEG polymer at a specific amino acid (BAY94-9027), has entered phase I clinical trials.12 In preclinical studies, this rFVIII variant showed reduced immunogenicity in animal models, potentially due to the ability of the PEG polymer to shield antigenic FVIII epitopes from immune effector cells.37
Glyco-PEGylation
Glyco-PEGylation of rFVIIa has been accomplished by Ghosh et al,38 and the modified protein showed similar amidolytic activity, slightly reduced cleavage of FX at lower concentrations (but not higher concentrations) in the presence of tissue factor, and reduced interaction with phospholipid. A phase 1/2 study of long-acting rFVIIa in patients with hemophilia A and B and inhibitors has now been completed14 ; analysis of the data is pending. In addition, a phase 1 clinical trial of a 40-kDa glyco-PEGylated rFIX has now been completed.13 In FIX-knockout mice, this modified rFIX exhibited equivalent potency to a commercially available rFIX and better duration of hemostasis after a bleeding challenge.39
A slightly different approach to PEGylation of coagulation proteins has been implemented involving association with PEGylated liposomes (PEGLip). Yatuv et al40 demonstrated noncovalent binding of rFVIIa with high specificity and affinity to PEGLip, and in severe hemophilic plasma containing anti-FVIII antibodies, faster clot formation and higher clot stability by thromboelastography (TEG). In rats, an extended circulation time and reduced clearance (1.4 times slower than free rVIIa) were observed with PEGLip-rVIIa, suggesting decreased clearance. However, despite promising preclinical41 and phase 1 data,42 a phase 2 clinical trial of rFVIII conjugated to PEGLip was terminated prematurely due to lack of efficacy. In that study, the PEGLip-rFVIII did not appear to circulate longer than non-PEGLip-rFVIII, and transient mild elevations in total and low density lipoprotein concentrations occurred.43 Not only recombinant, but also plasma-derived FVIII has been conjugated to PEGLip, with notably improved hemostatic activity in an animal model of hemophilia.44
Accumulation of PEG polymers has been documented in some (nonbleeding disorder) preclinical studies. Whether accumulation will be evident in patients receiving large doses of PEG-conjugated constructs over a long duration (ie, a lifetime) is not known. In addition, a substantial number of individuals exhibit anti-PEG antibodies, which may result in rapid clearance of PEG-protein conjugates,45,46 but data in hemophilia applications are absent.
Polysialylation
In contradistinction to polymers of PEG, which can be branched or linear and are not naturally occurring, polysialic acids are nonbranched polymers of N-acetylneuraminic acid that are found in a large array of mammalian tissues45 and therefore are unlikely to be immunogenic in humans. Polysialic acids can provide similar benefits to PEGylation if conjugated to therapeutic proteins.45 Although no trials of polysialylated therapeutic proteins have been performed in humans, in a FVIII-VWF double-knockout mouse, the half-life of polysialylated rFVIII was 4-fold that of rFVIII.15 Conjugation of polysialic acids may provide an alternative to PEG in application to other coagulation proteins as well.
Fusion to protein conjugates
Fc-IgG fusion
In recognition of the very long half-life of IgG molecules in the circulation, fusion of a single molecule of rFIX to the Fc-portion of IgG has been explored as a means to increase the circulation time of recombinant coagulation factors. The approach capitalizes on the serial reuptake of IgG into endothelial cells lining the vasculature and binding the neonatal Fc receptor (FcRn) found ubiquitously within. Such binding “recycles” the IgG molecule (and the fused protein) back to the cell surface and averts lysosomal degradation.47 In animal models of hemophilia, FIXFc fusion proteins exhibited an extended half-life of up to ∼ 48 hours in cynomolgus monkeys and hemophilia B dogs compared with standard rFIX (half-life ∼ 18 hours),6 and showed normal procoagulant activity in hemophilia B mice.17 The molecule is currently being evaluated in a phase 2/3 clinical trial in individuals with severe hemophilia,18 as is an analogously prepared rFVIII Fc construct.16
Fusion to albumin
Albumin-bound proteins are also recycled from the intravascular space by the FcRn, and a rFIX molecule fused to recombinant albumin via a cleavable linker peptide has been prepared.48 The molecule has shown comparable coagulant activity in a mouse model of hemophilia B, and extended circulatory half-life in other mammals.49 A phase 1/2 clinical trial in patients with severe hemophilia is under way.20 In rats, an albumin-fused rFVIIa molecule has exhibited a 6- to 7-fold increase in circulating half-life compared with unmodified rFVIIa, with preserved hemostatic efficacy.19
Site-directed mutagenesis
As mentioned previously, the introduction of point mutations to modify the amino acid sequence of a coagulation factor has allowed for targeted attachment of PEG polymers, resulting in a fully functional molecule with an extended half-life.36 Other modified rFVIII peptides, which have not yet been tested in humans, include an activated protein C–resistant rFVIII (IR8) with an A2 domain that is not susceptible to dissociation after activation, allowing for prolonged procoagulant activity,21 and engineered molecules in which disulfide bonds have been introduced to prevent decay by subunit dissociation and/or to increase potency.22,50
An rFIX mutein featuring an alanine in place of an arginine at residue 338 (FIX-R338A), introduced by a point mutation, was shown in vitro to yield 3-fold catalytic activity, probably due to alteration of FX binding.23 Such an increase in specific activity could diminish the amount of infused FIX required to treat or prevent bleeding episodes, or even allow for an enhanced response in gene therapy protocols.24 Similarly, a FIX-FX hybrid has been constructed by incorporating 4 active-site components of FX into a recombinant FIX molecule, leading to dramatically enhanced procoagulant activity.25
In a phase 2 dose-escalation trial, the modified rFVIIa molecule NN1731 (produced by the introduction of 3 point mutations at residues 158, 296, and 298, leading to an increase in tissue factor–independent catalytic activity) was compared in double-blind, randomized fashion with standard rVIIa for the treatment of bleeding episodes in individuals with severe hemophilia A or B and an inhibitor.27 Adverse events were similar between the groups, no immunogenicity was detected, and initial efficacy appeared to be very good in the groups treated with NN1731, but further studies are required to confirm a benefit over the currently available product. An engineered rFVIIa variant designed to exhibit tissue factor–independent activation of FIX and FX (BAY 86-6150) has been evaluated in a phase 1, randomized, double-blind, placebo-controlled, single-dose escalation study.26 In this trial, which enrolled adult men with moderate/severe hemophilia with or without inhibitors, the modified rFVIIa molecule showed a prolonged half-life of 5-7 hours compared with standard rFVIIa. No increase in analytes suggestive of a prothrombotic physiology (eg, D-dimers and thrombin-antithrombin complexes) was observed.
Further clinical trials are needed to confirm the clinical benefit and safety of genetically modified recombinant coagulation factors.
Alternative hemostatic approaches
Opportunities to enhance hemostasis in individuals with hemophilia (including those with inhibitors) lie not only in bioengineered coagulation factors, but also in a variety of novel, nonpeptide approaches that may help to support hemostasis and reduce dependence on the administration of replacement coagulation factors (Table 3). In addition, the following therapies have the potential for nontraditional (eg, oral or subcutaneous) administration, which could revolutionize the treatment paradigm and greatly enhance quality of life.
Aptamers
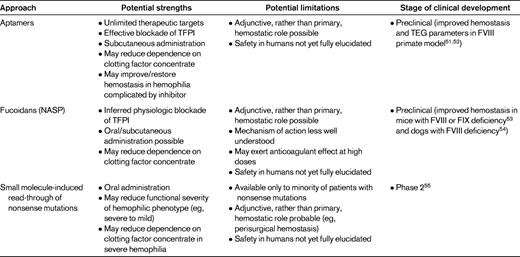
Aptamers, ribosomal or deoxyribosomal oligonucleic acids that theoretically can be produced to be of any length or 3-dimensional conformation, have properties that allow for binding to an almost unlimited array of physiologic targets. Although they were first developed for cancer therapeutics or antiviral treatment,56 they can also interact with hemostatic proteins. For example, inhibitory aptamers to coagulation serine proteases have been developed, and activation of FXII, kallikrein, and prothrombin by single-stranded aptamers has been described.57 Waters et al51 demonstrated hemostatic activity of aptamer ARC19499 in vitro by calibrated automated thrombogram to measure peak thrombin and endogenous thrombin potential. Both were reduced in hemophilia A plasma and hemophilia A plasma with inhibitors, but the aptamer, in a dose-dependent fashion and in similar manner to rVIIa, improved thrombin parameters in the coagulopathic plasmas. Both rVIIa and ARC19499 were able to restore clot time (R values) and development (angle) by TEG in hemophilia A plasma and in a nonhuman primate model (cynomolgus monkeys that had received anti-FVIII antibody). Treatment with the aptamer restored prolonged R values similar to values before anti-FVIII antibody treatment. As described in a later report,52 ARC19499 restored thrombin generation in hemophilia A and B plasmas, and at a 30nM concentration was found to have an effect equivalent to 14% FVIII activity in hemophilia A plasma. In addition, correction of bleeding time in a nonhuman primate saphenous vein model was observed after treatment with anti-FVIII antibody. Binding experiments using tissue factor pathway inhibitor (TFPI)–depleted plasma suggested that the hemostatic activity was dependent on interaction with TFPI. Another recent report58 has described the identification of an aptamer with activity against activated protein C, which could potentially allow for improved hemostasis in patients with hemophilia by virtue of therapeutic protein C inhibition, as has been described physiologically in children with severe hemophilia A with coinheritance of the factor V Leiden mutation.59
NASPs
Non-anticoagulant-sulfated polysaccharides (NASPs; also known as fucoidans) are heterogeneously sized, strongly anionic compounds with a fucose backbone that derive from marine plant sources (brown seaweed). NASPs have been investigated primarily for pharmacologic activity in anticancer, antiinflammation, antiangiogenesis, and even anticoagulant systems60,61 ; selected NASPs may enhance hemostasis. In 2008, Prasad et al54 described oral administration of AV513, a NASP with anti-TFPI activity,53 to severe hemophilia A dogs in a dose-escalating fashion. All 3 treated dogs showed an impressive improvement in TEG R value at the highest dose (20 mg/kg), but only 2 of the dogs showed normalization of a cuticle bleeding time. The animal who had not shown a reduction in cuticle bleeding time at the highest dose did show an improvement at a slightly lower dose (15 mg/kg), suggesting that interanimal dose-responsiveness may vary. The reason for this finding is not clear, but at high doses, AV513 had been shown to exhibit anticoagulant properties.53 These results suggest a potentially broad therapeutic range of NASPs in patients with hemophilia, and also encourage careful monitoring if the compounds reach human clinical trials, given the potential for dose-dependent anticoagulant activity.
Clinical studies are required to determine whether the chronic use of aptamers/NASPs might be able to modify a severe hemophilic phenotype to a degree similar to, and with the benefits of, the use of prophylactic infusions of clotting factor concentrate.
Read-through of nonsense mutations
Nonsense mutations, which involve substitution of a wild-type amino acid to lead to an in-frame UAA, UAG, or UGA codon, cause nonsense-mediated mRNA decay and premature termination of mRNA translation, producing destabilized mRNA and truncated (often nonfunctional) proteins.62 Approximately 10%-12%% of patients with hemophilia A are estimated to carry nonsense alleles, and the frequency is slightly higher among patients with hemophilia B.63,64 Modifying the rate of mRNA decay by interfering with factors that promote the destabilization of the nonsense mRNA could allow for the production of substantial biologically active protein.
In stable cell lines harboring nonsense mutations, the small molecule PTC124 (an oxidiazole linked to flurobenzene and benzoic acid rings) promoted dose-dependent read-through of UGA nonsense mutations.62 In transgenic mice expressing the R338X nonsense mutation leading to severe hemophilia B (FIX < 1 ng/mL), subcutaneous administration of PTC124 led to physiologically meaningful increases in FIX concentration (3-5 ng/mL) in a subset of the mice.65 PTC124 is orally bioavailable and nontoxic in healthy volunteers.66 Human trials in Duchenne muscular dystrophy and cystic fibrosis characterized by nonsense mutations are ongoing or have been completed, and a phase 2 trial in patients with stop codon–mediated severe hemophilia A or B is now under way.55 Insights from this clinical study may help to determine whether the suppression of nonsense mutations is a viable option for selected individuals with severe hemophilia, because it theoretically could lead to independence from clotting factor concentrate by virtue of conversion of the hemophilic phenotype from severe to moderate or mild.
Application of novel therapeutics to current clinical issues in hemophilia
Ultimately, any advance in medical therapy for hemophilia must address an unmet need. The following areas stand to gain the most from the aforementioned emerging therapeutic approaches, which may enable enduring rather than episodic or age-specific (ie, during the pediatric and adolescent years alone) correction of the hemophilic bleeding risk (Table 4).
Use of prophylaxis
Although the US Medical and Scientific Advisory Committee of the National Hemophilia Foundation has recommended thrice-weekly prophylaxis with clotting factor concentrate for children with severe hemophilia A beginning before the onset of frequent bleeding,67 a recent survey showed that half of US hemophilia treatment centers do not follow this recommendation, opting for less frequent infusions or later initiation of prophylaxis.68 Certainly, the requirement for frequent infusions mandating central venous access in very young children acts as a barrier to compliance with optimal care.1 In contrast, the benefit of continued prophylaxis into and throughout adulthood has not been proven conclusively, but the “standard” of on-demand treatment in response to bleeding episodes has also not been driven by data showing that it is a better approach than prophylaxis. In addition, the expense and inconvenience of frequent administrations of the currently available therapies are a deterrent.69 Because a significant proportion of adults with severe hemophilia currently find regular prophylactic infusions beneficial,69,70 longer-acting modified coagulation factors, those that can be administered more easily, or nonpeptide agents that enhance hemostasis may be all the more valuable.
Joint health
One of the major benefits of optimal use of prophylaxis is the prevention of repeated hemarthroses leading to disabling hemophilic arthropathy. Compared with a standard of frequent prophylactic infusions to keep the residual factor activity level > 1%,67 a suboptimal regimen of replacement of clotting factor concentrate is likely to be associated with worse joint outcomes in children.1 Recent data suggest that adults with severe hemophilia also maintain joint health when prophylaxis is initiated early and continued.71 Considering that an 18-year-old man with severe hemophilia A may endure over 5000 twice-weekly infusions of clotting factor concentrate if he wishes to maintain a prophylactic regimen over his lifetime, even a 50% reduction in infusion frequency by virtue of a longer-acting product could lead to greater compliance with enduring prophylactic regimens and may have a meaningful impact on musculoskeletal health.
HRQoL
Data regarding health-related quality-of-life (HRQoL) outcomes in patients of all ages with severe hemophilia are accumulating. Because of both decreased restrictions in mobility due to joint bleeding and increased physical activity, including the ability to participate in athletics,3 HRQoL measures improve in children whose severe hemophilic phenotype is modified by prophylactic infusions of clotting factor concentrate.72 Even in patients with hemophilia complicated by an inhibitor, HRQoL improved compared with baseline when secondary prophylaxis with a bypassing agent was administered, and the benefit continued in the postprophylaxis period.73 The potential impact of improved hemostasis therapies in susceptible hemophilic individuals on these and other HRQoL variables, such as missed days from work or school, is substantial.
Aging and hemostatic challenges
Increasing age exposes an individual to medical conditions (eg, cardiovascular disease or cancer) for which diagnosis and/or treatment involves the application of antithrombotic therapies or invasive procedures. As more individuals with congenital bleeding disorders advance in age, special needs for effective, durable, and well-tolerated correction of the coagulopathic defect will likely increase.4 In particular, intensive prophylaxis may be required for adults with moderate or severe hemophilia who require treatment with therapeutic-intensity anticoagulants or antiplatelet agents for ischemic cardiovascular disease74 or who require diagnostic or therapeutic surgery for cancer.4 Improved therapies therefore must address the emerging need for effective and well-tolerated hemostasis protocols specifically to support age-related medical or surgical bleeding challenges in persons with hemophilia.
Conclusion
Although great strides have been made in the treatment of hemophilia, an understanding of both the inherent limitations of currently available therapies and the unmet clinical needs of the affected population has provided a strong rationale for the development of newer drugs. These agents, which include both bioengineered coagulation factors and nonprotein therapeutics, hold promise for individuals with congenital deficiencies of FVIII and FIX and for those with inhibitors. Insights from clinical trials using these agents have the potential to modify current treatment paradigms and help patients enjoy improved joint health, HRQoL, and tolerability of hemostatic challenges.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from NovoNordisk, Bayer Healthcare, Baxter Healthcare, Biogen IDEC, and PTC Biotherapeutics and has received honoraria from NovoNordisk, Bayer Healthcare, and Baxter Healthcare. Off-label drug use: None disclosed.
Correspondence
Patrick F. Fogarty, MD, Penn Comprehensive Hemophilia and Thrombosis Program, Division of Hematology/Oncology, University of Pennsylvania, 3400 Spruce Street, 3 Dulles, Philadelphia, PA 19104; Phone: (215) 615-6555; Fax: (215) 615-6594; e-mail: patrick.fogarty@uphs.upenn.edu.