Abstract
Since the isolation and characterization of the genes for FVIII and FIX some 30 years ago, a longstanding goal of the field has been development of successful gene therapy for the hemophilias. In a landmark study published in 2011, Nathwani et al demonstrated successful conversion of severe hemophilia B to mild or moderate disease in 6 adult males who underwent intravenous infusion of an adeno-associated viral (AAV) vector expressing factor IX. These 6 subjects have now exhibited expression of FIX at levels ranging from 1% to 6% of normal for periods of > 2 years. This review discusses obstacles that were overcome to reach this goal and the next steps in clinical investigation. Safety issues that will need to be addressed before more widespread use of this approach are discussed. Efforts to extend AAV-mediated gene therapy to hemophilia A, and alternate approaches that may be useful for persons with severe liver disease, who may not be candidates for gene transfer to liver, are also discussed.
Introduction
As presented last year at the plenary session of ASH1 and published simultaneously in the New England Journal of Medicine,2 the longstanding goal of continuous expression of a clotting factor gene after a single administration of a viral vector carrying that gene has now been achieved in men with severe hemophilia B. The most obvious question that follows is how quickly, or whether, this can become a widely available treatment option for those with hemophilia, and if so, how it will fit into the evolving therapeutic landscape of the disease, as the long-acting clotting factor concentrates enter the market.3,4 Other questions (whether alternate approaches to gene therapy for hemophilia will also move forward, whether any of these strategies will be useful for people with liver disease or inhibitors, and whether gene therapy will be deemed safe for the pediatric population) also arise. This review provides a summary of recent laboratory and clinical investigation into these questions.
Current status of the field of gene therapy
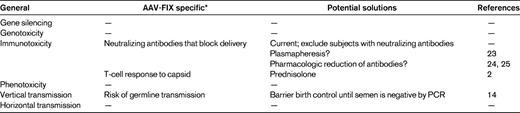
An area of major interest to both physicians and patients is when a gene therapy product for hemophilia might be available. A major obstacle to an accurate prediction of the timing of licensing is that there are no licensed gene therapy products in Europe or the United States at this time. Thus, it is not easy to draw a straight line from proof-of-concept studies in humans, which have now occurred, to the licensing of a product. However, what is clear is that clinical successes using gene therapy approaches have now been reported in a variety of disease states (eg, Leber congenital amaurosis5–8 ; ADA-SCID9 ; thalassemia10 ). These successes, as well as some highly visible complications, have helped to define the major safety issues that will need to be addressed to allow widespread use of any gene therapy product. The potential complications of gene therapy were predicted before clinical trials began (Table 1); experience has shaped the problem list, which differs depending on vector type and target tissue, and clinical and laboratory investigations have provided solutions, as has been the case for other classes of therapeutics. The next part of this review will summarize the obstacles encountered on the way to achieving long-term expression at therapeutic levels, and the solutions that have been developed.
Current status of gene therapy for hemophilia
The most promising clinical results in hemophilia currently are for hemophilia B, where intravenous infusion of an adeno-associated viral (AAV) vector encoding factor IX (FIX) under the control of a liver-restricted promoter has resulted in expression of FIX at plateau levels ranging from 1% to 6%, for periods of > 2 years, in 6 adult males with severe hemophilia B.2 Four of these 6 subjects, who were enrolled in the United Kingdom where twice-weekly prophylactic infusion of FIX concentrates is standard of care, have now been able to discontinue routine prophylaxis, reserving factor infusion for surgery or trauma, and the other 2 have been able to reduce the frequency of factor infusions. Those who stopped prophylaxis have largely been free of spontaneous bleeding episodes, confirming what had been predicted based on results in hemophilic dogs treated with the same approach,11 and on the natural history of mildly affected hemophilia patients. The safety of the approach to date has been excellent, with the only adverse event related to the study agent being a rise in liver enzymes (aspartate aminotransferase and alanine aminotransferase), accompanied by a decline in FIX levels, which resolved after a course of tapering steroids (see “Advances in the second AAV-FIX trial” for additional details).
Problems identified in the first AAV-FIX trial
When the “cure” of hemophilia B in the canine model of the disease was presented at the plenary session of ASH in 1998,12 most expected that the proof of principle in humans would follow quickly. However, providing abundant evidence for the observation that not all toxicities of a drug can be accurately predicted by studies in animals, the first clinical trial of hepatic artery infusion of an AAV2-FIX vector quickly encountered obstacles that had not been identified in the preclinical studies.13,14 The first of these was the appearance of vector sequences in DNA extracted from the semen of subjects enrolled on the trial. The so-called risk of vertical transmission (Table 1) obviously does not pose any sort of threat to the patient himself but rather represents a risk for potential offspring if the spermatocytes are transduced. The theoretical concern is that, if a genetically modified germ cell fertilized an egg, the inserted DNA could alter the exquisitely timed series of gene expression/repression events that characterize normal embryonic/fetal development. The trial was placed on hold for 9 months as more data were developed in an animal model that is frequently used for reproductive toxicology studies, the rabbit.15 These data suggested that the presence of vector DNA in the semen was transient and that the risk of observing this was dose-related.16,17 The trial was therefore modified to include a recommendation to bank sperm in advance of the procedure for those considering fathering additional children, and a recommendation that participants use barrier birth control until 2 sequential semen specimens were negative by PCR for vector sequences (Table 1). Subsequent clinical studies have confirmed the animal data, and there have been no cases of semen being persistently positive for vector sequences after administration of either AAV2 or AAV8 vectors.2,14
Another general class of obstacles encountered was that of humoral and cellular immunity to AAV, a consequence of the fact that AAV vectors have been engineered from a wild-type parvovirus naturally infecting humans.18–20 Neutralizing antibodies to AAV, which are present in a substantial proportion (likely 30%-50%) of the population,19,21 do not block administration of vector directly into a target tissue (eg, into muscle by intramuscular injection,22 or into the retina by subretinal injection8 ) but are clearly an obstacle when vector is delivered through the circulation. The direct comparison of 2 subjects injected at the high dose in this first trial showed that a neutralizing antibody titer of 1:17 effectively blocked transduction (Figure 1A-B), and subsequent trials have restricted subject enrollment to those with low or undetectable neutralizing antibodies against AAV (Table 2). This requirement bars a substantial fraction of subjects from participation in the ongoing trials. Proposed solutions to this problem have included plasmapheresis23 or drugs that target B cells, but early studies suggest that these will have limited effects against high titers of neutralizing antibodies.24,25

Results in AAV-mediated gene therapy trials for subjects infused at a dose of 2 × 1012 vg/kg (high dose in both trials). (A-B) Of the 2 subjects infused at the highest dose with the AAV2-FIX vector, the first achieved a circulating level of ∼ 10% initially, followed by a gradual fall to the baseline of < 1%. This was accompanied by an asymptomatic rise in liver enzymes that began 4 weeks after vector infusion. The second subject, with a higher pretreatment neutralizing antibody titer to AAV-2, failed to achieve any substantial expression. (C-D) The 2 subjects infused at the highest dose with the AAV8-scFIX vector both achieved circulating levels in the range of 6%-10% initially. (C) The first showed a rise in liver enzymes and fall in FIX levels ∼ 8 weeks after vector infusion, but administration of prednisolone at 60 mg/d normalized liver function tests and arrested the decline in FIX levels, leaving the subject with sustained expression in the range of 2%. (D) In the second subject infused at this dose, liver function tests rose very slightly ∼ 9 weeks after vector infusion, the subject was immediately started on prednisolone, and his circulating FIX level now remains in the range of 5%.2 (C-D) Reprinted from Figure 1E and F in Nathwani et al2 with permission.
Results in AAV-mediated gene therapy trials for subjects infused at a dose of 2 × 1012 vg/kg (high dose in both trials). (A-B) Of the 2 subjects infused at the highest dose with the AAV2-FIX vector, the first achieved a circulating level of ∼ 10% initially, followed by a gradual fall to the baseline of < 1%. This was accompanied by an asymptomatic rise in liver enzymes that began 4 weeks after vector infusion. The second subject, with a higher pretreatment neutralizing antibody titer to AAV-2, failed to achieve any substantial expression. (C-D) The 2 subjects infused at the highest dose with the AAV8-scFIX vector both achieved circulating levels in the range of 6%-10% initially. (C) The first showed a rise in liver enzymes and fall in FIX levels ∼ 8 weeks after vector infusion, but administration of prednisolone at 60 mg/d normalized liver function tests and arrested the decline in FIX levels, leaving the subject with sustained expression in the range of 2%. (D) In the second subject infused at this dose, liver function tests rose very slightly ∼ 9 weeks after vector infusion, the subject was immediately started on prednisolone, and his circulating FIX level now remains in the range of 5%.2 (C-D) Reprinted from Figure 1E and F in Nathwani et al2 with permission.
The second immune obstacle identified in the AAV2-FIX trial was what appears to be a memory T-cell response to AAV, that if unchecked, can result in immune-mediated destruction of the transduced hepatocytes and thus loss of circulating levels of FIX. The initial presentation of this phenomenon, which had not been seen in animal models, was in a subject that received the highest dose of vector, 2 × 1012 vg/kg. Beginning 4 weeks after vector infusion, the FIX levels in the circulation, which had risen to 10%-12%, began to fall gradually, reaching the pretreatment baseline of < 1% at 12 weeks after infusion (Figure 1A). Simultaneously, the alanine aminotransferase and aspartate aminotransferase, which had remained in the normal range initially, began to rise, peaking at 5 weeks at levels of 532 and 202 IU/L, respectively, and then slowly returning to baseline with no medical intervention. The subject felt well during this time and maintained his normal activity levels. Thus, the event was self-limited and asymptomatic; and despite the loss of expression of the donated gene, the subject continued to respond well to recombinant FIX concentrates with no evidence of inhibitor formation. Data from a subsequent subject enrolled in the study suggested that vector infusion triggered expansion of a clone of AAV capsid-specific CD8+ T cells, which targeted transduced hepatocytes that presented capsid-derived peptides via MHC class I, resulting in a loss of FIX expression and a transient and self-limited rise in transaminases (Figure 2). These results suggested that some sort of immunomodulatory regimen might be needed to achieve long-term therapeutic efficacy in humans after AAV-FIX gene transfer.

Hypothesis explaining simultaneous rise in liver enzymes and decline in FIX levels at high dose in AAV-FIX trials. (A) AAV enters the cell via receptor-mediated uptake into clathrin-coated endosomes. (B) On endosomal escape, AAV vector is released into the cytosol. Vector genomes are then released from the capsid in a process called uncoating. (C) At least some of the capsid undergoes ubiquitination and proteasomal processing. Capsid-derived peptides are transported to the endoplasmic reticulum and loaded onto MHC class I molecules. The peptide/MHC complexes are displayed on the surface of the transduced cell. (D) MHC class I presentation flags hepatocytes for recognition by capsid-specific CD8+ T cells, resulting in release of cytolytic granules and clearance of the transduced cells. (E) Vector is also taken up by antigen-presenting cells and presented via MHC class II, resulting in (F) activation of CD4+ T helper cells and production of cytokines. Adapted with permission from Figure 3 in Mingozzi and High.61
Hypothesis explaining simultaneous rise in liver enzymes and decline in FIX levels at high dose in AAV-FIX trials. (A) AAV enters the cell via receptor-mediated uptake into clathrin-coated endosomes. (B) On endosomal escape, AAV vector is released into the cytosol. Vector genomes are then released from the capsid in a process called uncoating. (C) At least some of the capsid undergoes ubiquitination and proteasomal processing. Capsid-derived peptides are transported to the endoplasmic reticulum and loaded onto MHC class I molecules. The peptide/MHC complexes are displayed on the surface of the transduced cell. (D) MHC class I presentation flags hepatocytes for recognition by capsid-specific CD8+ T cells, resulting in release of cytolytic granules and clearance of the transduced cells. (E) Vector is also taken up by antigen-presenting cells and presented via MHC class II, resulting in (F) activation of CD4+ T helper cells and production of cytokines. Adapted with permission from Figure 3 in Mingozzi and High.61
Advances in the second AAV-FIX trial
Two critical advances were achieved in the second AAV-FIX trial. Based on the discovery that alternate serotypes of AAV displayed higher tropism for liver compared with AAV2, Nathwani et al developed an AAV8-FIX vector and demonstrated that therapeutically relevant levels of transduction could be achieved with intravenous infusion as opposed to the hepatic artery infusion that had been used in the first trial, making the vector delivery procedure much simpler and more appealing. Other modifications aimed at enhancing the potency of the vector included a self-complementary genome and codon optimization.26,27 Based on observations from the first trial, the AAV8-FIX trial also incorporated a provision to start subjects on a course of high-dose steroids if liver enzymes rose or if FIX levels began to fall. The trial began in March 2010 as a dose escalation study that would include 2 subjects at each of 3 dose cohorts ranging from 2 × 1011 vg/kg to 2 × 1012 vg/kg; vector infusion was uneventful, and all subjects were discharged after being observed in hospital overnight. This trial excluded subjects with detectable neutralizing antibodies to AAV8. The first 4 subjects all demonstrated a modest improvement in circulating FIX levels, in the range of 1%-3%, but clearly different from the pretreatment values.2 The fifth subject, infused at a dose of 2 × 1012 vg/kg, initially demonstrated FIX levels in the range of 8%-10% of normal, but at ∼ 8 weeks after infusion, the transgene expression level dropped and the liver enzymes rose (alanine aminotransferase and aspartate aminotransferase to 202 IU/L and 143 IU/L, respectively). The subject was started on a course of prednisolone (60 mg/d), which was tapered slowly over the ensuing 8 weeks. IFN-γ ELISPOT studies to monitor for T-cell responses against AAV capsid, FIX peptides, and potentially translated alternate open reading frames within the FIX sequence showed only a response to capsid, as had been demonstrated in the previous trial. The institution of steroid therapy was associated with a rapid reduction in capsid-specific T cells in PBMCs, resolution of the elevated transaminases, and eventual stabilization of the FIX levels at 2% of normal (Figure 1C). The sixth subject, at about the same time point, 8-9 weeks after vector infusion, also showed a very slight rise (still within normal limits) in serum transaminases and a decline in the FIX level. He was immediately started on prednisolone, which was tapered over a 4-week period, and remains with an FIX level of 6%, more than one year after the steroids were tapered (Figure 1D). Thus, for the subjects in the first 2 dose cohorts, improvement in FIX levels was modest, in the range of 1%-3%, but they did not require prednisolone and the levels have been stable, whereas at least one of the subjects in the high-dose cohort has achieved a better plateau level, ∼ 6% (enough to convert hemophilia from severe to mild) but required a short course of prednisolone.
Clearly, the ability to infuse vector intravenously rather than through the hepatic artery in an invasive procedure in the radiology suite and the discovery that the CD8+ T-cell response could be reduced or controlled by a course of steroids were major steps forward in the development of an AAV-FIX therapy. Current efforts have been focused on extending these results; the study of larger numbers of subjects should allow investigators to determine the proportion of adults that are likely to require coadministration of immunomodulatory drugs, and whether prednisolone will reliably control the immune response if it is encountered. Additional AAV-FIX trials are now underway or getting started, with most being some variant of the already-conducted study.28 Multiple trials should allow more rapid accumulation of data, and perhaps extension to groups of subjects who are currently excluded (eg, those who are HIV-positive, hepatitis C virus (HCV) RNA viral load-positive, and those with neutralizing antibodies to AAV; Table 2).
It is worth noting that there has been no evidence of inhibitors to FIX in any of the AAV-FIX trials. Most trials require that persons have at least 20 exposure days to FIX protein concentrates and no history of an inhibitor, but there are no exclusions based on the underlying mutation.
The interplay of manufacturing issues with toxicities related to the human immune response
Viral vectors are arguably one of the most complex therapeutics yet manufactured because they consist of a nucleic acid sequence (the active agent), contained within a highly ordered set of proteins (the viral capsid), which serve to guide the DNA to the target cell. Some of the complexities encountered in clinical development proceed from the fact that the viral vector is engineered from a naturally occurring virus and therefore has the capacity to trigger memory responses on exposure to the human immune system. These immune responses arose originally in response to a virus, which can replicate (in the presence of helper virus, such as adenovirus) but can be recalled by the vector, which cannot replicate and indeed is not a virus. Although it is hardly surprising that the immune response does not initially distinguish between these 2 entities, the clinician needs to manage these responses to achieve the best outcome for the patient. This is, in effect, a new area of clinical therapeutics, and approaching it rationally is further complicated by the absence of a reliable animal model of the human cellular immune response to AAV vectors.
A byproduct of most AAV manufacturing procedures is a large excess of empty capsids (ie, fully assembled capsids that are devoid of any packaged DNA). In many manufacturing procedures, these can constitute a majority of the initial product (> 90%).29–31 Manufacturing processes vary in terms of whether these “empties” are removed or not. Evidence has accumulated, though,32 to suggest that empty AAV capsids can gain access to a target cell and thus contribute to the intracellular capsid burden that will eventually be processed and presented on the surface of the transduced cell, flagging these cells for destruction by capsid-specific CD8+ T cells. The logical implication is that removing empty capsids from AAV vector preparations would be beneficial because they do not add to the therapeutic effect of the product and may contribute to toxicity. The vector preparation used in the AAV8-FIX trial sponsored by St Jude Children's Research Hospital and University College London was ∼ 80% empties.29 Thus, it may be of interest to study subjects infused at the same dose but using a vector preparation from which empties have been removed. One could hypothesize that reducing the total capsid burden may allow administration of the top dose previously used, 2 × 1012 vg/kg, without requiring a course of steroids. This in turn might allow inclusion of subjects who are HCV RNA viral load-positive, who are now generally excluded, because high-dose steroids must be avoided in this population. Another strategy that may allow investigators to avoid steroids is to use a higher specific activity variant of FIX, such as FIX Padua.33 Studies in hemophilic dogs have shown that the use of a FIX Padua transgene can result in activity levels of up to 40% with FIX antigen levels of only 4%.34 The risk here is that the non–wild-type transgene product may be immunogenic, but work in a number of animal models has established that expression of any transgene from an AAV vector in the liver tends to promote tolerance to the transgene product,35 so this is probably relatively low risk.
Remaining safety issues
Although the total number of subjects injected with AAV-FIX vectors targeting the liver is small (13 have been reported in the literature to date2,14 ), the short-term safety issues have probably been delineated and consist of the risk of germline transmission, and the risk of transaminase elevation associated with loss of expression of the donated gene (Table 1). The latter appears to be more of an efficacy “risk” than a true safety risk because the transaminase elevation and accompanying decline in FIX levels have to date been asymptomatic and self-limited. The long-term risks of AAV transduction of human liver are more difficult to assess. Although AAV vectors are predominantly nonintegrating, with most of the expression coming from stable episomes,36,37 direct sequencing of DNA extracted from target organs in animals has shown that integration can occur.37,38 A single report in which mice with β-glucuronidase deficiency and their healthy littermates were injected with an AAV2 vector as neonates described an increase in hepatocellular carcinoma in mice > 13 months of age39 (an advanced age for mice), and a subsequent study showed that some of the tumors contained integrated vector DNA within a 6-kb locus rich in regulatory RNAs of unknown function.40 Another study in which mice were followed for 18 months failed to demonstrate a significantly higher rate of hepatocellular carcinoma compared with saline-injected controls,41 and there have been no reports of hepatocellular carcinoma in large animals or humans exposed to AAV. More investigation on mechanism will be required to determine whether the observed integration events can bring about transformation and to assess the level of risk that this represents for human subjects receiving AAV.
Potential applicability to hemophilia A
The use of an AAV vector to direct expression of FVIII in mice was first reported by Burton et al in 1999.42 To circumvent the limitations imposed by the limited genome packaging capacity of AAV vectors (∼ 5 kb is the maximum size of a single-stranded DNA fragment that can be accommodated within the AAV capsid), they used 2 vectors: one encoding the heavy chain and the other the light chain. Subsequently Chao et al43 reported the use of a single vector expressing B domain-deleted FVIII to correct the bleeding diathesis in hemophilia A mice. The published data in large animal models, which have been a better guide to dosing in humans than have mouse data, required doses on the order of 0.6-4 × 1013 vg/kg to achieve circulating levels of canine FVIII in the range of 1%-7.8%.44,45 These doses are at least a log higher than doses already administered in humans with AAV-FIX vectors (maximum dose thus far is 2 × 1012 vg/kg). Whether the immune response triggered by a dose in the range of 2 × 1013 vg/kg can be successfully managed by a course of steroids is still unknown. Recently, however, McVey et al described a codon-optimized human FVIII construct that, in the context of a lentiviral vector, directed FVIII expression levels that were 29- to 44-fold higher than those directed by non–codon-optimized constructs.46 If this level of improvement in expression is preserved in an AAV vector, this may reduce the dose required for therapeutic efficacy to a range that has already been safely tested in humans and thus permit initiation of AAV-FVIII trials.
Alternate approaches
The safety of AAV-mediated gene transfer to liver in persons with severe liver disease has not been established, although the AAV2 trial did include 3 persons with mild liver abnormalities (HCV RNA viral load-positive, and Metavir score of F1 on liver biopsy) who were infused without incident.14 Because most trials will now include a provision for high-dose steroids if a rise in liver enzymes occurs, it will be difficult to include those who are HCV RNA viral load-positive. Strategies that target other organs, including megakaryocytes,47,48 the joints themselves,49 and skeletal muscle,50,51 have been proposed, and the latter has undergone safety testing in clinical trials.22,52 Transduction of hematopoietic cells with retroviral or lentiviral vectors expressing FVIII under the control of a platelet-specific promoter, followed by transplantation of the autologous gene-modified cells, has been carried out successfully in hemophilic mice53 and has been shown to effect hemostasis even in mice with inhibitors.54 Detailed analysis of the platelets has shown that the FVIII is stored in platelet α-granules and released at the site of injury when platelets are activated. However, studies in mice suggest that both spatial and temporal aspects of clot formation may differ based on whether FVIII is supplied from plasma versus a gene-modified platelet48 ; this aspect may require more extensive preclinical analysis.
Another class of persons for whom AAV-mediated gene transfer to liver may have limited success are young children. Because AAV vectors are predominantly nonintegrating, expression is gradually lost if vector is injected into a growing animal. Alternative strategies are under investigation, including lentiviral transduction of liver,55,56 the use of transposase-transposon technology to integrate the donated gene into the host genome,57 and the use of zinc finger nucleases to effect genome editing.58 These would be predicted to install a permanent change or correction into the genome, which would be passed to daughter cells, and thus expected to mediate long-term expression even if administered early in life. Published studies with these methods, however, have not progressed beyond mouse data at this writing.
In conclusion, although the number of subjects is small, the landmark success reported by Nathwani et al provides clear proof of concept that AAV-mediated, liver-directed gene transfer can provide long-term expression of FIX at levels adequate to convert severe hemophilia B to mild disease. At this point, this demonstration has occurred in a relatively restricted subset of the hemophilia patient population: adults, who are HIV-negative, HCV RNA viral load-negative, and lack neutralizing antibodies to AAV8. Clinical studies over the next few years will determine whether this patient group can be safely and effectively expanded and whether the same approach will be effective for hemophilia A. The journey from proof of concept in humans to commercial availability can be long: in a recent article, Dr Ulla Hedner described the 14-year effort from the first convincing use of purified FVIIa in a patient with an inhibitor (in 1982), to the licensing of the recombinant product in Europe (1996).59 On the other hand, under the pressure of the HIV epidemic, recombinant FVIII went from first infusion in a human subject (1987) to a licensed product (1992) in just 5 years.60 The pace of product development in gene therapy remains to be seen. But the first and most critical step of the journey has now been accomplished.
This article was selected by the Blood and Hematology 2012 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2012. This article is reprinted with permission from Blood. 2012; Volume 120.
Disclosures
Conflict-of-interest disclosure: K.A.H. is an Investigator of the Howard Hughes Medical Institute that supported this work. This work was also supported in part by the National Institutes of Health National Heart, Lung, and Blood Institute (P01 HL64190 and P01 HL078810). K.A.H. holds patents in the area of AAV-FIX; she has waived all financial interest in these. K.A.H. is also a coinventor on patents related to AAV manufacture and purification, and lentiviral vector manufacture; she holds equity in bluebird bio Inc, which has clinical programs in X-linked adrenoleukodystrophy and thalassemia; and she has served as a consultant to BioMarin Pharmaceuticals, Biogen Idec, Genzyme, Novo Nordisk, Nordic Biotech Advisors, Pfizer, and Shire regarding gene therapy programs. Off-label drug use: Prednisone, immunomodulation.
Correspondence
Katherine A. High, 3501 Civic Center Blvd, Rm 5060 CTRB, The Children's Hospital of Philadelphia, Philadelphia, PA 19104; Phone: 215-590-4521; Fax: 215-590-3660; e-mail: high@email.chop.edu.