Abstract
Genomic profiling has transformed our understanding of the genetic basis of acute lymphoblastic leukemia (ALL). Recent years have seen a shift from microarray analysis and candidate gene sequencing to next-generation sequencing. Together, these approaches have shown that many ALL subtypes are characterized by constellations of structural rearrangements, submicroscopic DNA copy number alterations, and sequence mutations, several of which have clear implications for risk stratification and targeted therapeutic intervention. Mutations in genes regulating lymphoid development are a hallmark of ALL, and alterations of the lymphoid transcription factor gene IKZF1 (IKAROS) are associated with a high risk of treatment failure in B-ALL. Approximately 20% of B-ALL cases harbor genetic alterations that activate kinase signaling that may be amenable to treatment with tyrosine kinase inhibitors, including rearrangements of the cytokine receptor gene CRLF2; rearrangements of ABL1, JAK2, and PDGFRB; and mutations of JAK1 and JAK2. Whole-genome sequencing has also identified novel targets of mutation in aggressive T-lineage ALL, including hematopoietic regulators (ETV6 and RUNX1), tyrosine kinases, and epigenetic regulators. Challenges for the future are to comprehensively identify and experimentally validate all genetic alterations driving leukemogenesis and treatment failure in childhood and adult ALL and to implement genomic profiling into the clinical setting to guide risk stratification and targeted therapy.
The genetic landscape of ALL
Acute lymphoblastic leukemia (ALL) is the most common childhood tumor,1 and although more than 80% of children are cured, relapsed ALL remains a leading cause of childhood morbidity and mortality. With increasing age, the frequency of genetic alterations associated with favorable outcome declines and alterations associated with poor outcome such as BCR-ABL1 are more common. With the exception of tyrosine kinase inhibitors (TKIs) such as imatinib in the treatment of BCR-ABL1+ leukemia, current therapies do not target specific genetic alterations and are associated with substantial short- and long-term toxicities that limit further dose escalation. There is therefore great interest in the use of high-resolution genomic profiling to characterize the genetic basis of leukemogenesis, to understand and predict treatment failure, and to provide novel markers that may be integrated into diagnostic testing and be targeted with novel therapies. These studies also provide critical insights into the nature of clonal heterogeneity and tumor evolution. Ongoing next-generation sequencing of ALL has already provided important insights in acute leukemia not evident from “presequencing” genetic profiling approaches. Recent insights obtained from genomic profiling of ALL, with an emphasis on high-risk B-progenitor ALL, are reviewed herein.
Approximately 75% of childhood ALL cases harbor a recurring chromosomal alteration detectable by karyotyping, FISH, or molecular techniques (Figure 1). In B-progenitor ALL, these include hyperdiploidy with greater than 50 chromosomes, hypodiploidy with less than 44 chromosomes, and chromosomal rearrangements including t(12;21) ETV6-RUNX1 (TEL-AML1), t(1;19) TCF3-PBX1 (E2A-PBX1), t(9;22) BCR-ABL1, and rearrangement of MLL at 11q23 to a diverse range of fusion partners. T-lineage ALL is characterized by activating mutations of NOTCH1 and rearrangements of transcription factors TLX1 (HOX11), TLX3 (HOX11L2), LYL1, TAL1, and MLL.2 High hyperdiploidy, ETV6-RUNX1 (both associated with favorable outcome), and TCF3-PBX1 are less common in adult ALL. Although these rearrangements are important initiating events in leukemogenesis and are widely used in diagnosis and risk stratification algorithms, they are insufficient to fully explain leukemogenesis. Rearrangements such as ETV6-RUNX1 are present years before the development of leukemia, and many do not alone result in the development of leukemia in experimental models. It is now known that the majority of ALL cases are characterized by distinct constellations of structural and submicroscopic genetic alterations and sequence mutations.

Submicroscopic genetic alterations in ALL
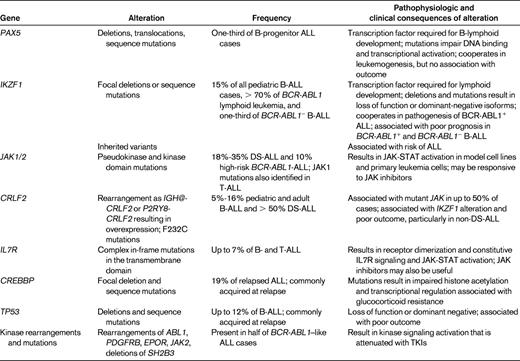
In 2007-2008, multiple studies described the genomic landscape of DNA copy number alterations in ALL using array-based comparative genomic hybridization or single nucleotide polymorphism (SNP) microarrays. The majority of these studies examined childhood ALL cohorts, and similarly detailed studies of the genomics of adolescent and adult ALL are ongoing but are currently incomplete. ALL genomes typically harbor fewer structural alterations than many solid tumors, but more than 50 recurring deletions or amplifications have been identified, many of which involve a single gene or few genes (reviewed in Mullighan and Downing3 ; Table 1). Many of the genes involved encode proteins with key roles in lymphoid development (eg, PAX5, IZKF1, EBF1, and LMO2), cell-cycle regulation and tumor suppression (CDKN2A/CDKN2B, PTEN, and RB1), lymphoid signaling (BTLA, CD200, TOX, and the glucocorticoid receptor NR3C1), and transcriptional regulation and coactivation (TBL1XR1, ETV6, and ERG; Figure 2). Several genes are involved in multiple types of genetic alteration, including copy number alteration translocation and sequence mutation, such as PAX5, WT1, and PTEN, indicating that microarray profiling is alone incapable of detecting all genetic alterations in ALL.

Proposed schema for the role of genetic alterations in the pathogenesis of B-ALL. It is likely that chromosomal rearrangements are acquired early in leukemogenesis and drive transcriptional and epigenetic dysregulation and aberrant self-renewal. These lesions and/or secondary genetic alterations disrupt lymphoid development and result in an arrest in maturation. Additional genetic alterations target multiple cellular pathways, including cell-cycle regulation, tumor suppression, and chromatin modification. In a subset of cases (BCR-ABL1+ and BCR-ABL1–like ALL), genetic alterations drive aberrant cytokine receptor and kinase signaling. Together, these events result in the proliferation and establishment of the leukemic clone. Diagnosis ALL samples are commonly clonally heterogeneous, and genetic alterations in minor clones may confer resistance to therapy and promote disease relapse. It should be noted that a direct role of many of the genetic alterations shown in the pathogenesis of ALL has not yet been confirmed experimentally. A similar schema can be proposed for T-ALL, in which lesions targeting lymphoid development, self-renewal, and kinase signaling are also observed and in which there are multiple targets of mutations with unknown roles in leukemogenesis (eg, PHF6 and WT1).
Proposed schema for the role of genetic alterations in the pathogenesis of B-ALL. It is likely that chromosomal rearrangements are acquired early in leukemogenesis and drive transcriptional and epigenetic dysregulation and aberrant self-renewal. These lesions and/or secondary genetic alterations disrupt lymphoid development and result in an arrest in maturation. Additional genetic alterations target multiple cellular pathways, including cell-cycle regulation, tumor suppression, and chromatin modification. In a subset of cases (BCR-ABL1+ and BCR-ABL1–like ALL), genetic alterations drive aberrant cytokine receptor and kinase signaling. Together, these events result in the proliferation and establishment of the leukemic clone. Diagnosis ALL samples are commonly clonally heterogeneous, and genetic alterations in minor clones may confer resistance to therapy and promote disease relapse. It should be noted that a direct role of many of the genetic alterations shown in the pathogenesis of ALL has not yet been confirmed experimentally. A similar schema can be proposed for T-ALL, in which lesions targeting lymphoid development, self-renewal, and kinase signaling are also observed and in which there are multiple targets of mutations with unknown roles in leukemogenesis (eg, PHF6 and WT1).
The nature and frequency of genetic lesions is subtype dependent. MLL-rearranged leukemias harbor very few additional structural or sequence alterations.4,5 In contrast, ETV6-RUNX1 and BCR-ABL1 ALL harbor more alterations. Emerging experimental data have shown that several of these alterations cooperate in leukemogenesis. Deletion of Pax5 and Ikzf1 accelerates the onset of leukemia in retroviral BM transplantation, transgenic models of BCR-ABL1 ALL, and chemical and retroviral models of leukemia.6
Although many of these alterations are enriched in specific cytogenetic ALL subtypes, a notable exception is alteration of the ETS-family transcription factor ERG (ETS-related gene), which occurs exclusively in patients lacking known chromosomal rearrangements and is a hallmark of a novel subtype of B-ALL with a distinct gene-expression profile and generally favorable outcome. The ERG deletions involve an internal subset of exons resulting in loss of the central inhibitory and pointed domains and expression of an aberrant C-terminal ERG fragment that retains the ETS and transactivation domains and functions as a competitive inhibitor of wild-type ERG.7
IKZF1 alterations in B-ALL
Relatively few of these novel genetic alterations have been found to be reproducibly associated with outcome, with the notable exception of alterations of the lymphoid transcription factor gene IKZF1 (IKAROS) in B-ALL. Alterations of the transcriptional regulation of lymphoid development is a hallmark of B-ALL, with deletion, sequence mutations, or rearrangements of the transcription factors PAX5, IKZF1, and EBF1 present in more than two-thirds of patients.4,8 PAX5 alterations are present in one-third of B-ALL patients, but are not associated with outcome.8,9 In contrast, IKZF1 deletions (and sequence mutations) are less common, being present in approximately 15% of childhood ALL cases and are a hallmark of 2 types of high-risk ALL. IKZF1 encodes IKAROS, the founding member of a family of zinc finger transcription factors that is required for the development of all lymphoid lineages. The IKZF1 alterations observed in ALL include focal or broad deletions that result in loss of function and internal deletions of coding exons 4-7 that remove the N-terminal DNA-binding zinc fingers, leading to expression of a dominant-negative isoform, IK6. IKZF1 alterations are present in more than 70% of BCR-ABL1 lymphoid leukemia, including de novo ALL and chronic myeloid leukemia at progression to lymphoid blast crisis,10 and are associated with poor outcome in BCR-ABL1+ ALL.11 Multiple studies have reported associations between alterations of IKZF1 and poor outcome in BCR-ABL1− ALL.8,12 Moreover, many of these BCR-ABL1−, IKZF1-mutated cases exhibit a gene-expression profile similar to BCR-ABL1+ ALL and harbor novel kinase-activating mutations and rearrangements (see “BCR-ABL–like ALL” below).
CRLF2 rearrangements and JAK mutations in ALL
FISH and SNP array profiling have identified CRLF2 rearrangements in approximately 7% of childhood ALL cases (Figure 1) and 50% of cases associated with Down syndrome (DS-ALL).13,14 CRLF2 is located in the pseudoautosomal region (PAR1) at Xp22.3/Yp11.3 and encodes cytokine receptor–like factor 2 (also known as the thymic stromal lymphopoietin receptor). With IL7 receptor alpha (IL7Rα), CRLF2 forms a heterodimeric receptor for thymic stromal lymphopoietin. CRLF2 is rearranged by translocation into the immunoglobulin heavy chain locus (IGH@-CRLF2) or, more commonly, by a focal deletion upstream of CRLF2 that results in expression of P2RY8-CRLF2 that encodes full-length CRLF2. Both rearrangements result in aberrant overexpression of CRLF2 on the cell surface of leukemic lymphoblasts that may be detected by immunophenotyping. Less commonly, a p.Phe232Cys CRLF2 mutation results in receptor overexpression.
CRLF2 alterations are associated with the presence of activating mutations in the JAK genes JAK1 and JAK2,13–15 which, with the exception of T-lineage ALL, are otherwise uncommon in ALL. The JAK family comprises 4 members, JAK1, JAK2, JAK3, and TYK2. The JAK/STAT pathway mediates signaling from cytokine, chemokine, and growth factor receptors via the JAK nonreceptor tyrosine kinases and the STAT family of transcription factors. The JAK mutations are most commonly missense mutations at R683 in the pseudokinase domain of JAK2 and are distinct from the JAK2 V617F mutations that are a hallmark of myeloproliferative diseases. Less common are activating mutations in the kinase domain of JAK1 and JAK2. Like the JAK2 V617F mutation, the JAK1/2 mutant alleles observed in ALL are transforming in vitro.16,17 Up to 50% of CRLF2-rearranged cases harbor activating JAK1/2 mutations, and conversely, almost all cases of B-ALL with JAK1/2 mutations harbor concomitant rearrangements of CRLF2. Coexpression of CRLF2 and JAK1/2 mutations is transforming in vitro, suggesting that these 2 lesions are central in lymphoid transformation. Half of CRLF2-rearranged cases lack a JAK mutation and the nature of alternative kinase signaling mutations in these cases is unknown. In non–DS-ALL, CRLF2 alterations and JAK mutations are associated with IKZF1 deletion/mutation, a gene-expression profile similar to BCR-ABL1+ ALL, and, in some studies, poor outcome.18 The associations between CRLF2 and outcome have varied between studies and cohorts in part due to differences in the cohorts studied.19–21 The association is most consistent in non–DS-ALL cohorts of high-risk B-progenitor ALL. However, it should be emphasized that few studies have performed comprehensive analysis of CRLF2 rearrangement and expression, JAK1 and JAK2 mutational screening (including both pseudokinase and kinase domain mutations), and detection of IKZF1 deletions and sequence mutations. Recent studies performed by the Children's Oncology Group (COG) have shown that CRLF2 and IKZF1 alterations are associated with inferior outcome in multiple cohorts and that elevated CRLF2 expression in the absence of rearrangement is also an adverse prognostic feature.22
Regardless of JAK mutation status, the leukemic cells harboring CRLF2 deregulation exhibit activation of JAK-STAT and PI3K/mTOR pathways and are sensitive to JAK and mTOR inhibitors in vitro and in vivo. An early-phase trial of the JAK inhibitor ruxolitinib in relapsed and refractory childhood tumors, including cases with CRLF2 rearrangement and/or JAK mutations, ADVL1011, has been initiated (www.clinicaltrials.gov identifier NCT01164163).
BCR-ABL1–like ALL
Up to 15% of childhood B-progenitor ALL patients lack a previously identified chromosomal rearrangement, but exhibit a gene-expression profile highly similar to that of BCR-ABL1+ ALL (reviewed by Dr Thomas in this section) and often have deletion/mutation of IKZF1, which is also common in BCR-ABL1-ALL (Figure 1).8,23 Ongoing studies suggest that BCR-ABL1–like ALL is also common in adolescent and young adult ALL (C.G.M., C. L. Willman, unpublished data). The prognosis of BCR-ABL1–like ALL is poor. In the COG AALL0232 study of high-risk B-progenitor ALL, the event-free survival for BCR-ABL1–like cases was 62.6% ± 6.9% compared with 85.8% ± 2.0% for non–BCR-ABL1–like cases (P < .0001) and was associated with poor outcome after adjustment for age, sex, peripheral blood leukocyte count at presentation, and day 29 flow cytometric levels of minimal residual disease.24 Up to half of BCR-ABL1–like cases harbor rearrangements of CRLF2 and JAK1/2 sequence mutations.17,18
To identify the genetic basis of non–CRLF2-rearranged BCR-ABL1–like ALL, the COG, as part of the US National Cancer Institute Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative (http://target.cancer.gov/) performed mRNA-seq and whole-genome sequencing (WGS) of diagnostic and matched remission DNA and/or RNA for 15 BCR-ABL1–like cases. This identified a range of novel rearrangements, copy number alterations, and sequence mutations activating kinase signaling, including rearrangements of PDGFRB, ABL1, JAK2, and EPOR, as well as deletion/mutation of SH2B3 and the IL7R.25 Several of these alterations confer growth factor independence in murine Ba/F3 and Arf−/− cell lines that is attenuated with currently available TKIs such as the ABL1/PDGFRB inhibitors imatinib, dasatinib, and dovitinib and the JAK2 inhibitor ruxolitinib. Moreover, primary leukemic cells from patients with BCR-ABL1–like ALL exhibit activation of these signaling pathways on phosphoflow cytometric signaling analysis, and xenograft studies have shown that the engrafted tumors are sensitive to appropriately targeted TKI therapy.26 These exciting genomic and preclinical findings strongly suggest that patients with BCR-ABL1–like ALL, many of whom are at high risk of relapse and fail current maximal therapy, may be successfully treated with TKIs. Furthermore, these data indicate that concomitant lesions that disrupt hematopoietic development and drive proliferation are common in ALL, as proposed previously for acute myeloid leukemia. Ongoing work is extending sequencing efforts to identify the full repertoire of kinase-activating lesions in childhood and adult ALL. In addition, because BCR-ABL1–like ALL is associated with a distinct gene-expression signature (with overexpression of genes including MUC4, PON2, IGJ, and GPR110),25 patients may be identified at diagnosis by targeted gene-expression profiling and/or phosphoflow cytometry followed by sequencing approaches.
Intrachromosomal amplification of chromosome 21
Intrachromosomal amplification of chromosome 21 (iAMP21) occurs in up to 2% of B-progenitor ALL patients, and in the United Kingdom childhood ALL trials has been associated with older age and poor outcome.27 iAMP 21 is defined by gain of at least 3 copies of the region of chromosome 21 containing RUNX1. The amplification is often large and complex and accompanied by deletion of the subtelomeric regions of chromosome 21. The basis of generation of iAMP21 and the mechanistic contribution of this abnormality to leukemogenesis are unclear, but identification may be performed to identify cases of ALL at increased risk of relapse.
Hypodiploid ALL
Hypodiploidy with less than 45 chromosomes is associated with a very high risk of treatment failure, the genetic basis of which has been poorly understood.28 Hypodiploid ALL may subclassified by degree of aneuploidy into near haploid (NH-ALL, 24-31 chromosomes) and low hypodiploid (LH-ALL, 32-44 chromosomes) cases. In conjunction with the COG, we performed a detailed genomic analysis of more than 120 hypodiploid ALL cases, including WGS or exome sequencing of more than 40 cases.29 This sequencing showed that NH-ALL has a very high frequency of deletions and sequence mutations that activate Ras signaling, including recurring novel alterations of NF1, and that NH- and LH-ALL have a high frequency of inactivating alterations of the IKAROS genes IKZF2 (HELIOS) and IKZF3 (AIOLOS) that are otherwise rare in ALL. With transcriptional profiling, these results indicate that NH- and LH-ALL are distinct diseases. Moreover, we demonstrated Ras pathway activation by biochemical and phosphosignaling analysis, suggesting that therapeutic targeting of this pathway may represent an important novel treatment outcome in this high-risk leukemia.
Sequence mutations in ALL
Candidate gene-sequencing studies, in which genes have been selected for sequencing based on a priori knowledge of the role of a gene product in lymphoid development, tumorigenesis, or the knowledge that a gene is involved by structural genetic alterations, have identified multiple targets of mutation in ALL (eg, PAX5, BCL11B, FBXW7, IKZF1, LEF1, WT1, PTEN1, and NF1). In general, these studies have shown that DNA copy number alterations are more common than sequence mutations in ALL; however, detailed analysis of sequence mutations in large cohorts of ALL has been prohibitively costly.
To characterize sequence variation in ALL and to further explore the genetic basis of relapse, we performed Sanger sequencing of 300 genes from 24 B- and T-lineage ALL patients at diagnosis, relapse, and remission.30 The frequency of sequence mutation was low (0-5 per case) and, as observed for DNA-copy number alterations, many deleterious mutations present at diagnosis were no longer evident at relapse, including mutations in the Ras signaling pathway (eg, NRAS, KRAS, PTPN11, and NF1) and B-cell development (eg, PAX5, but not IKZF1, deletions/mutations of which were always preserved at relapse or acquired as new lesions). Deletion or mutation of CREBBP, encoding the transcriptional coactivators and acetyltransferase CREB binding protein (also known as CBP), were present in almost 20% of relapsed ALL cases and appeared to be particularly enriched in relapsed hyperdiploid ALL, a subtype normally associated with favorable outcome.31 The mutations identified are enriched in the histone acetyltransferase (HAT) domain and attenuated the normal HAT activity of murine CREBBP. Moreover, CREBBP mediates the transcriptional response to glucocorticoid therapy and the mutations were shown to disrupt the normal transcriptional response to glucocorticoids. Therefore, CREBBP mutations may represent an important mechanism underlying treatment failure in ALL, and may be targeted with agents that modulate the level of histone acetylation in leukemic cells, such as histone deacetylase inhibitors. Recent studies have also shown that mutations in TP53 (p53), otherwise infrequent in ALL, are also enriched at relapse and associated with poor outcome.32
Recent insights into the genetic basis of T-lineage ALL
Several studies have used second-generation sequencing to identify novel genetic alterations in T-lineage ALL. T-ALL exhibits a peak in incidence in older male children, and to investigate this sex bias, Ferrando et al performed exon capture and sequencing of X-chromosome genes.33 This identified a high frequency of deleterious mutations in PHF6, which encodes PHD finger protein 6, a zinc finger–containing putative transcriptional factor. The role of PHF6 alterations in T-cell leukemogenesis is at present unclear.
In 2009, Campana et al at St Jude's Children's Research Hospital described the entity of “early T-cell precursor” ALL (ETP-ALL), in which leukemic blasts exhibit expression of cytoplasmic CD3, but no expression of CD1a and CD8, weak or absent expression of CD5, aberrant expression of stem cell and myeloid markers, and immunophenotypic and transcriptional similarity to the murine early T-cell precursor that retains developmental plasticity.34 Multiple studies have shown that ETP-ALL comprises 10%-15% of childhood and adult ALL, although diagnostic immunophenotypic criteria have varied between studies. ETP-ALL is associated with poor treatment response, induction failure, and poor event-free survival. ETP-ALL blasts lack alterations otherwise common in T-ALL, such as activating NOTCH1 mutations and CDKN2A/B deletions, but lack common chromosomal rearrangement.
The St Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project performed WGS of tumor and matched nontumor DNA from 12 children with ETP-ALL and examined the frequency of novel and recurring genetic alterations in an additional 54 ETP- and 42 non–ETP-ALL patients.35 This showed a marked diversity in the nature and frequency of somatic sequence and structural alterations and showed that 3 pathways are commonly mutated in ETP-ALL: hematopoietic development, Ras and/or cytokine receptor/JAK-STAT signaling, and histone modification. Several other studies have also identified mutations in several of these genes and pathways using non-WGS approaches.36 Fifty-eight percent of ETP cases (compared with 17% of non-ETP T-ALL cases) harbored loss-of-function or dominant-negative mutations in developmental genes, including RUNX1, IKZF1, ETV6, GATA3, and EP300. Several of these genes are known targets of mutation in hematopoietic malignancies (eg, RUNX1 in myeloid disorders and ETV6 and IKZF1 in B-progenitor ALL), but have not previously been implicated in T-ALL. Activating signaling mutations were identified in 67% of cases (compared with 19% of non-ETP T-ALL cases), including mutations in NRAS, KRAS, JAK1, NF1, and PTPN11 and novel mutations in JAK3, SH2B3 (encoding LNK, a negative regulator of JAK2 signaling), and IL7R. Several studies have now identified mutations in IL7R in both T-lineage and B-progenitor ALL.37 The IL7R mutations are located in the transmembrane domain and commonly introduce a cysteine that results in receptor dimerization and constitutive JAK-STAT that is abrogated by pharmacologic JAK inhibitors such as ruxolitinib.
An unexpected finding was a high frequency of mutations in epigenetic regulators in ETP-ALL, the most frequent of which were components of the polycomb repressor complex 2 (PRC2), a H3K27 trimethylase that normally induces transcriptional repression and antagonizes the transcriptional activating effects of MLL. The most commonly mutated gene was EZH2, which encodes the catalytic component of the complex. EZH2 is also mutated in follicular lymphoma, but, in contrast to the highly recurrent Y641 mutations observed in FL that are gain of function,38 the mutations in T-ALL occur in other sites in EZH2 and are predicted to disrupt the catalytic SET domain and result in loss of function.
These pathways—hematopoietic development, signaling, and epigenetic regulation—are also frequently mutated in AML. In addition, the transcriptional profile of ETP-ALL is highly similar to that of normal hematopoietic stem cells, and that of high risk AML but not the normal human ETP. This suggests that ETP-ALL may represent a stem cell or progenitor leukemia, and recent data examining biphenotypic leukemias have identified similar mutations in a small number of cases,39 suggesting that the entity of ETP-ALL may extend beyond leukemias nominally of T-cell (eg, cCD3+) lineage. Moreover, these findings suggest that non-ALL regimens, whether myeloid directed, targeted, or epigenetic therapies, should be pursued in this disease.
Clonal heterogeneity in ALL
Cytogenetic analysis demonstrated many years ago that genetic alterations in leukemic cells may change during therapy. SNP array and mutational profiling of serial ALL samples has confirmed that the majority of ALL cases exhibit substantial changes in genetic alterations from diagnosis to relapse. Relapse in ALL commonly arises from the emergence of a minor subclone that shares some genetic features and also harbors distinct genetic alterations from the predominant clone at diagnosis. Less commonly, the predominant clones at diagnosis or relapse are identical or share no commonality. Therefore, at diagnosis, individual patients harbor multiple genetically distinct clones that share a common clonal origin, which then respond differently to the selective pressure of antileukemic therapy. This hypothesis is supported by recent xenograft models of ALL comparing the genomic alterations in engrafted tumors with the primary sample and tracing the clonal evolution of tumors.40 This has important clinical implications, because among the lesions that may not be detected at diagnosis by profiling of the by leukemia clone yet emerge at relapse are alterations of IKZF1. Therefore, whereas many patients with IKZF1 alteration at high risk of relapse may be identified by genomic profiling of bulk leukemic samples by standard approaches, others will require highly sensitive methods, for example, quantitative DNA or RNA PCR, to identify these alterations (which may be deletions or mutations) at very low levels at the time of diagnosis. Moreover, the full range of genetic alterations contributing to relapse remains to be defined.
Conclusions and future directions
It is likely that current genome-sequencing efforts will identify all somatic genetic alterations present in ALL genomes within the next few years. These studies are currently most advanced for childhood B-progenitor ALL, and it will be crucial to carefully compare the nature and frequency of genetic alterations between childhood and adult ALL. At present, these studies are focused on characterizing somatic genetic alterations in coding genes, but it will also be important to identify of all inherited variations contributing to leukemogenesis and to determine the role of noncoding genomic variation in leukemia development.
Several genetic alterations are of clear clinical importance for risk stratification or therapeutic targeting, including detection of IKZF1 alterations and identification of patients with kinase-activating alterations that may benefit from targeted TKI therapy. Identification of all such alterations is not trivial, because multiple types of mutations affect these genes. Currently, clinical next-generation sequencing poses substantial logistic challenges and, although not widely available at present, is likely will become increasingly commonplace in the next few years. In the interim, multiple groups are pursuing focused genetic testing of individual lesions and screening approaches to identify pathway dysregulation, such as flow cytometric assays to identify activated kinase signaling in B-ALL. It is likely that these approaches will improve current risk stratification and ultimately improve outcome for patients with high-risk ALL.
Acknowledgments
The author thanks members of his laboratory and colleagues at St Jude Children's Research Hospital, the Children's Oncology Group, and the National Cancer Institute TARGET consortium, whose efforts contributed to the work described in this review, and apologizes to those whose work could not be described or cited due to space constraints.
This research was supported by the American Lebanese Syrian Associated Charities of St Jude Children's Research Hospital, the National Cancer Institute of the US National Institutes of Health, the Pew Charitable Trusts, the American Society of Hematology, the American Association for Cancer Research, Stand Up To Cancer, the St Baldrick's Foundation, the National Health and Medical Research Council (Australia), and the Swedish Research Council.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Use of TKIs in ALL patients with novel kinase activation alterations.
Correspondence
Charles G. Mullighan, Department of Pathology, St Jude Children's Research Hospital, 262 Danny Thomas Pl, Mail Stop 342, Rm 4047G, Memphis, TN 38105; Phone: 901-595-3387; Fax: 901-595-5947; e-mail: charles.mullighan@stjude.org.