Abstract
The myelodysplastic syndromes (MDS) are a clinically and cytogenetically heterogeneous group of clonal diseases characterized by ineffective hematopoiesis, peripheral blood cytopenias, and an increased risk of progression to acute myeloid leukemia. The precise molecular mechanisms behind the development of MDS have remained elusive; however, the distinct sensitivity of this disease to DNA methyltransferase inhibitors and the presence of markedly abnormal epigenetic profiles suggested the existence of an epigenetic mechanism underlying the disease. Recently, the advent of new technologies for the detection of genetic abnormalities has led to the description of a set of novel recurrent mutations in patients with this disease. The majority of these novel mutations have been described in genes encoding different components of the epigenetic machinery, many of which are associated with distinct clinical outcomes. Finally, mutations in mRNA splicing genes have also been described recently in MDS, underscoring the molecular complexity that underlies the development of this heterogeneous disease.
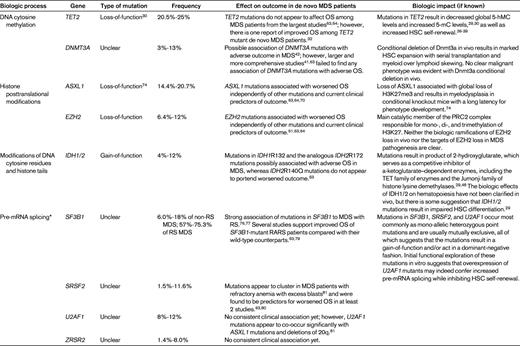
The myelodysplastic syndromes (MDS) are a group of clonal disorders of the hematopoietic system characterized by the presence of ineffective hematopoiesis, peripheral cytopenias, and an increased risk of transformation to acute myeloid leukemia (AML). Clinically and cytogenetically heterogeneous, the molecular mechanisms behind MDS have been difficult to identify. The observation that conventional cytogenetics fails to reveal clonal cytogenetic abnormalities at diagnosis in approximately half of MDS cases, coupled with the disease's distinct sensitivity to DNA methyltransferase inhibitors, led to the hypothesis that epigenetic alterations were critical in the pathogenesis of these disorders. Several DNA methylation profiling studies have since confirmed the presence of a profoundly abnormal epigenome in this disease1,2 and in multiple human malignancies.3,4 However, the use of more sensitive genetic techniques in recent years has led to the discovery of a higher incidence of genetic abnormalities in MDS than those detected by standard cytogenetic analysis. Most of these novel mutations in MDS target different components of the epigenetic machinery, suggesting a link between the presence of these mutations and the epigenetic abnormalities observed in this disease. More recently, somatic mutations in the spliceosome machinery have also been described. This class of mutations appears to be uniquely enriched in patients with MDS compared with patients with other myeloid malignancies not characterized by dysplasia. Herein we review the most prominent findings from recent years in the molecular genetics of MDS and their impact on disease biology (Table 1).
The epigenetic nature of MDS
Before genome-wide epigenetic studies were feasible, aberrant silencing of tumor-suppressor and DNA damage-repair genes through hypermethylation of promoter-associated CpG islands was described in MDS.5–8 The advent of genome-wide epigenetic studies allowed for a more comprehensive study of the epigenome in this disease. Studies by several groups revealed the presence of marked DNA methylation abnormalities consisting of profound hypermethylation.1,2 Whereas MDS is particularly resistant to conventional chemotherapy, treatment with cytosine analog DNA methyltransferase (DNMT) inhibitors such as 5-azacytidine (AZA) and decitabine improves hematopoiesis, increases overall survival (OS) and delays the onset of AML progression.9–12 However, to date, there has been very limited success in establishing a correlation between these abnormal epigenetic profiles and the response to DNMT inhibitor therapy1,13,14 raising the question of whether these aberrant epigenetic profiles are the initiating mechanism in MDS or if they are simply downstream consequences of other pathogenic mechanisms. Whereas functional studies demonstrating the role of DNA methylation in establishing the MDS clone have not been reported as yet, 2 different groups have reported impairment of leukemogenesis in the context of a Dnmt1 hypomorph murine model,15,16 indicating that DNA methylation is required for full malignant transformation in AML.
The DNA methylation machinery
In mammals, DNA methylation occurs exclusively at the 5′ position of cytosine nucleotides in the context of a CpG dinucleotide. The addition of the methyl group is catalyzed by the DNMT family of proteins, DNMT3A, DNMT3B and DNMT1 (reviewed in Herman and Baylin17 ). DNMT3A is a member of the mammalian family of DNA methyltransferases that add a methyl group to cytosine enzymatically in CpG dinucleotides. DNMT3A and its homolog, DNMT3B, are responsible for initiating de novo DNA methylation. In contrast, DNMT1 maintains methylation through cell division. DNMT3A also forms complexes with DNMT3L, a DNMT family member that lacks a methyltransferase catalytic domain; formation of the DNMT3A-DNMT3L complex results in increased DNMT3A activity. DNMT3L binds to histone H3 tails in the absence of histone H3 lysine 4 methylation, leading to the recruitment of DNMT3A activity to specific loci.18
Removal of the methyl group from the 5′ position of cytosine nucleotides was originally believed to be a passive mechanism achieved through subsequent DNA replication cycles. However, the recent discovery of the function of the Ten-Eleven Translocation (TET) proteins TET1, TET2 and TET3 has resulted in the identification of additional active mechanisms of DNA demethylation. TET proteins are alpha-ketoglutarate (aKG)– and Fe II–dependent oxygenases capable of modifying 5-methyl-cytosine (5mC) to generate 5-OH-methlcytosine (5hmC).19 The aKG required for this reaction is produced by Isocitrate Dehydrogenase 1 (IDH1) and IDH2 from isocitrate. 5hmC can then undergo further modification in a series of intermediary steps that are still not fully established, but which appear to involve the base excision repair mechanism that replaces this modified cytosine with an unmethylated cytosine, resulting in active DNA demethylation.20–22 Mutations in every component of the DNA machinery have been described in MDS and in de novo AML (Figure 1).

Recurrent somatic mutations affecting genes involved in epigenetic regulation of transcription in patients with MDS. Shown are mutations in proteins and complexes affecting (1) histone posttranslational modifications (PTMs); (2) DNA cytosine methylation, hydroxymethylation and demethylation; and (3) mRNA splicing. Copy-number loss and loss-of-function mutations affecting the PRC2 members EZH2, EED, and SUZ12 have been identified recurrently in patients with MDS, and mutations in EZH2 appear to be important predictors of worsened OS in patients with de novo MDS. The PRC2 complex serves to place 1-3 methyl groups on the 27th lysine residue of histone H3 (H3K27). Mutations in ASXL1 are among the most common mutations in patients with MDS, and these mutations have been shown recently to be loss-of-function mutations also associated with loss of H3K27me3. In addition to histone PTMs, mutations affecting DNA cytosine modifications have also been identified in MDS patients, including mutations of unclear function in the de novo DNA cytosine methyltransferase DNMT3A and loss-of-function mutations in the hydroxymethyltransferase TET2. Although the function of hydroxymethylcytosine is not yet totally clear, it is thought to represent an intermediate step in the demethylation of DNA. More recently, mutations in multiple genes encoding members of the spliceosome have also been identified in patients with MDS, including mutations of unclear function in SF3B1, U2AF1, SRFS2, and ZRSR2.
Recurrent somatic mutations affecting genes involved in epigenetic regulation of transcription in patients with MDS. Shown are mutations in proteins and complexes affecting (1) histone posttranslational modifications (PTMs); (2) DNA cytosine methylation, hydroxymethylation and demethylation; and (3) mRNA splicing. Copy-number loss and loss-of-function mutations affecting the PRC2 members EZH2, EED, and SUZ12 have been identified recurrently in patients with MDS, and mutations in EZH2 appear to be important predictors of worsened OS in patients with de novo MDS. The PRC2 complex serves to place 1-3 methyl groups on the 27th lysine residue of histone H3 (H3K27). Mutations in ASXL1 are among the most common mutations in patients with MDS, and these mutations have been shown recently to be loss-of-function mutations also associated with loss of H3K27me3. In addition to histone PTMs, mutations affecting DNA cytosine modifications have also been identified in MDS patients, including mutations of unclear function in the de novo DNA cytosine methyltransferase DNMT3A and loss-of-function mutations in the hydroxymethyltransferase TET2. Although the function of hydroxymethylcytosine is not yet totally clear, it is thought to represent an intermediate step in the demethylation of DNA. More recently, mutations in multiple genes encoding members of the spliceosome have also been identified in patients with MDS, including mutations of unclear function in SF3B1, U2AF1, SRFS2, and ZRSR2.
TET2
Whereas the TET1 gene had been described as an MLL fusion partner in t(10;11)(q22;q23) AML in 2003,23 it was not until 2009 that mutations in the TET2 gene, located on chromosome 4q24, were identified in MDS and other myeloid malignancies.24 Abnormalities in this gene are observed in 19%-26% of MDS patients, but can be as high as 58% in MDS/myeloproliferative neoplasm (MPN) overlap syndromes25,26 and include deletions, loss of heterozygosity, missense, nonsense, and frameshift mutations, resulting in impaired TET2 function.24–28
Given its role in initiating active DNA demethylation, decreased function in TET2 would be predicted to result in an increase in DNA methylation; however, studies examining genome-wide DNA methylation patterns in MDS and AML have reported both hypermethylated and hypomethylated profiles associated with the presence of TET2 mutations.29,30 Whether these discrepancies stem from technical differences in the platforms and cell fractions used in these studies or if they are related to the different disease context in which they were performed (AML vs MDS) or the presence of additional mutations in other genes in the different cohorts is yet to be determined. However, despite these differences, a consistent finding of decreased 5hmC levels in TET2-mutated patients has been reported by these groups,30 whereas an increase in overall 5mC levels was reported in these patients by other groups.31
The clinical impact of TET2 mutations in MDS is also a matter of debate. Whereas initial studies reported a favorable outcome for TET2-mutated patients,32 later, larger studies failed to confirm this initial report.33,34 More recently, The French MDS Study Group (GFM) reported that the presence of TET2 mutations was associated with an increased overall response rate to AZA treatment (82% vs 45% in mutant and wild-type TET2, respectively, P < .007). Moreover, TET2 mutation status in this study was an independent predictor of the overall response rate to AZA (odds ratio = 5.92; 95% confidence interval, 1.43-24.39; P = .014). However, this was not associated with a significant difference in either response duration or OS.35
Recently, several different models of Tet2 deletion have been reported in vivo.36–39 In all of these studies, deletion of Tet2 was associated with a remarkable increase in hematopoietic stem cell (HSC) number and self-renewal, a finding seen even with deletion of a single copy of Tet2. In addition, conditional or constitutional Tet2 deletion appears to result in a leukocytosis due to monocytosis and marked splenomegaly with a latency of at least 3 months.
DNMT3A
Recurrent mutations in the de novo DNA methyltransferase DNMT3A were first reported in AML in 2010,40 and were subsequently described in MDS, in which they occur at a much lower frequency (3%-8%)41,42 (Table 1). DNMT3A mutations in MDS are associated with older age at diagnosis, but do not associate significantly with any other clinical, morphologic, or cytogenetic feature of the disease.42,43 The low frequency of these mutations, along with the relatively small size of the cohorts studied, has hindered any attempts at determining the clinical impact of these mutations in this disease; however, univariate analysis in these studies revealed a worse clinical outcome in the mutant DNMT3A patients, with lower OS and faster progression to AML.42,43
Mutations in DNMT3A are heterozygous, and although they have been reported in almost every part of the open reading frame of DNMT3A, the majority of these mutations are missense mutations that cluster at the methyltransferase (MTase) domain, where they target highly conserved residues.42 The exact functional consequences of the different mutations has not been established yet, and although the frequently occurring R882 mutant that targets the MTase domain has been shown in vitro to have decreased methyltransferase activity,44 the functional consequences of this and other MTase domain mutations in the presence of the wild-type allele have not been reported to date. In patient specimens, total cytosine methylation levels remain unchanged40,41 and a limited genome-wide microarray analysis of 5 mutant AML patients revealed 182 loci that were hypomethylated compared with the DNMT3A wild-type controls.40 However, none of these studies stratified their analysis by the presence of coexistent mutations and it is too early to tell whether the impact in overall DNA methylation levels or distribution patterns is affected by the presence of other mutations (especially those mutations that target other epigenetic modifiers).
The presence of DNMT3A mutations in nearly all cells in the BM of MDS patients, irrespective of blast count, has led to the belief that this is an early genetic event in the development of this disease that may result in a clonal advantage of cells harboring the mutation.42 Murine models expressing mutant Dnmt3a are being pursued by several groups, however, the results from these studies have not been reported yet. Recently, conditional silencing of Dnmt3a in the hematopoietic system led to progressive impairment of HSC differentiation with simultaneous expansion of the HSC compartment. This murine model therefore demonstrated that Dnmt3a is essential for normal HSC differentiation and plays a critical role in the silencing of HSC-regulatory genes.45
IDH1 and IDH2
Isocitrate dehydrogenase 1 (IDH1) and IDH2 are metabolic enzymes that catalyze the conversion of isocitrate to aKG in an NADP+-dependent manner. Missense mutations in these genes occur at specific conserved residues (R132 in IDH1; R140 and R172 in IDH2) and are always heterozygous. These mutations result in a change in substrate specificity, so that mutant IDH1/2 enzymes are no longer capable of converting isocitrate into aKG, but instead use aKG as a substrate and catalyze its conversion into 2-hydroxyglutarate (2-HG) in a reaction that consumes NADPH.46,47 2-HG, in turn, inhibits TET proteins directly, thus linking mutations in these metabolic enzymes to the epigenetic machinery.48 Inhibition of TET proteins by 2-HG leads to a reduction in 5hmC levels with a concomitant increase in 5mC levels and the presence of widespread promoter hypermethylation29,48 (Figure 2). 2-HG can also inhibit other aKG-dependent epigenetic modifiers, such as the Jumonji-C domain histone demethylases49,50 ; however, the impact of IDH1/2 mutations on histone methylation in MDS has not been established yet. Finally, it has been shown recently that the (R)-enantiomer of 2-HG may actually promote the activity of the aKG-dependent EGLN hypoxia-inducible family of prolyl hydroxylases.51 The relevance of this gain-of-function activity induced by 2-HG to MDS pathogenesis has yet to be clarified.

Mutations in IDH1/2 result in 2-HG production and inhibition of TET2 function. Mutant IDH1 and IDH2 use the aKG produced by their wild-type counterpart as a substrate to produce the oncometabolite 2-HG, which in turns acts as a competitive inhibitor of TET2, thereby blocking the DNA hydroxymethylation pathway, resulting in an increase in the levels of methylated cytosine.
Mutations in IDH1/2 result in 2-HG production and inhibition of TET2 function. Mutant IDH1 and IDH2 use the aKG produced by their wild-type counterpart as a substrate to produce the oncometabolite 2-HG, which in turns acts as a competitive inhibitor of TET2, thereby blocking the DNA hydroxymethylation pathway, resulting in an increase in the levels of methylated cytosine.
Mutations in IDH1 were first described in gliomas and later discovered to be recurrent in approximately 8% of AML patients.52 The detection of 2-HG in AML patients who were wild-type for IDH1 led to the discovery of IDH2 mutations in AML and other myeloid malignancies.47 IDH1 and 2 mutations in MDS are seen in 4%-12% of cases53–56 (Table 1). Initial studies reported an unfavorable prognosis for MDS patients harboring mutations in IDH1, resulting in decreased OS and an increased risk of transformation to AML.56 Conversely, a study in which both IDH1 and IDH2 mutations were considered did not find any significant differences in clinical outcome in mutated patients.55 More recently, a large study reported by the Mayo Clinic showed that IDH2 R140 mutations were not associated with poor survival or increased risk of transformation to AML, whereas the other IDH1 and IDH2 mutations retained their negative impact on clinical outcome.53 Similar findings have also been reported recently in de novo AML.57
Histone-modifying enzymes
Epigenetic regulation does not occur at the DNA methylation level only—it is also dependent on the presence of a series of posttranslational modifications to the histone proteins at specific residues, including acetylation, methylation, and ubiquitination. The addition of these posttranslational modifications to the histone proteins is catalyzed by a group of histone-modifying enzymes with distinct specificity, a subset of which have been found to be mutated in MDS (Figure 1).
EZH2 and PRC2
Enhancer of Zeste Homolog 2 (EZH2) is the main catalytic member of the Polycomb Repressive Complex 2 (PRC2), which is responsible for mono-, di-, and trimethylation of lysine 27 on histone 3 (H3K27me1, me2, and me3, respectively), histone modifications strongly associated with transcriptional repression (Figure 1). Although increased activity of EZH2 due to overexpression has been demonstrated to be important in the pathogenesis of several epithelial malignancies58,59 and gain-of-function mutations in EZH2 have been identified in diffuse large B-cell lymphomas,60 in 2010, 2 independent groups discovered somatic loss-of-function mutations in EZH2 in patients with MDS, MDS/MPN overlap syndromes, and primary myelofibrosis61,62 (Table 1). From this work and subsequent studies, it is now clear that mutations in EZH2 occur in 6.4%-12% of patients with de novo MDS and appear to be biomarkers of worsened outcome in this group.61,63,64 Furthermore, in a comprehensive sequencing effort of 439 patients with de novo MDS, Bejar et al demonstrated that mutations in EZH2 appear to be associated with worsened OS in MDS patients independently of other known predictors of outcome such as the International Prognostic Scoring System value and other mutations.64 Curiously, mutations in EZH2 appear to be extraordinarily rare among patients with de novo AML (0 of 398 patients),57 suggesting that these mutations may be an important hallmark of hematopoietic malignancies marked by dysplasia. Further studies to validate the rarity of mutations in EZH2 in de novo AML are needed. In addition to somatic loss-of-function mutations in EZH2, rare additional deletions and putative loss-of-function mutations have been identified in the other core PRC2 members in MDS patients, including SUZ12 and EED mutations (all at less than 5% frequency).65,66
The discovery of mutations in PRC2 components in patients with MDS has led to several questions of key biologic and clinical interest. First, clear targets of EZH2 loss have yet to be identified in patients with MDS and animal models of conditional Ezh2 deletion have actually resulted in a phenotype of T-cell acute lymphoblastic leukemia (another human malignancy recently discovered to have PRC2 loss-of-function alterations) without a clear myeloid phenotype.67 Secondly, the discovery of EZH2 loss-of-function mutations in patients with MDS has led to the concern about potential unwanted side effects of therapies in early clinical development targeting EZH2 in the treatment of epithelial malignancies and large cell lymphomas.
ASXL1
Mutations in Addition of Sex Combs-like 1 (ASXL1) were originally identified by Gelsi-Boyer et al through candidate gene-sequencing studies of genes located in a focal deletion of 20q11 in MDS patient samples.68 Subsequent studies have found that mutations in ASXL1 are among the most common mutations in patients with MDS, occurring in 14.4%-20.7% of MDS patients64,68–70 and in 38%-43% of patients with MDS/MPN overlap syndromes68,71 (Table 1). Recently, mutations in all ASXL family members (ASXL1-3) have been identified in 2%-10% of patients with metastatic castration-resistant prostate cancer.72
Similar to EZH2 mutations, mutations in ASXL1 have been found to be associated consistently with worsened OS among patients with MDS independently of other clinical parameters, including age, cytogenetics, and number of cytopenias.63,64,70 Mutations in ASXL1 occur as somatic nonsense, out-of-frame insertion/deletions and nonsense mutations all in the 5′ end of the last exon.71
The conspicuous distribution of ASXL1 mutations led to question of whether these mutations actually represent loss-of-function or gain-of-function alterations. Biochemical studies of ASX, the ASXL1 homolog in Drosophila, suggested that ASXL1 may serve in a biochemical complex with at least one additional partner (termed BAP1 in mammals) to antagonize the PRC1 complex and remove a mono-ubiquitination from histone H2A lysine 119.73 Based on this work, loss of ASXL1 might be expected to result in an increase in H2AK119Ub, a mark associated with transcriptional repression. However, recent work suggests that ASXL1 mutations are bona fide loss-of-function mutations that are associated strongly with a loss of the transcriptionally repressive mark H3K27me3 and with minimal global effects on H2AK119Ub in myeloid hematopoietic cells.74 Intersection of the distribution of H3K27me3 with gene-expression profiling in isogenic cells with and without ASXL1 loss resulted in the identification of several targets of ASXL1, including up-regulation of the HOXA cluster of genes with ASXL1 loss.
In terms of understanding the potential biologic ramifications of ASXL1 loss in vivo, Fisher et al reported the phenotype of mice with constitutional deletion of Asxl1 in a gene-trap murine model shortly after the discovery of ASXL1 mutations in MDS patients.75 Deletion of Asxl1 in this manner resulted in a significant perinatal lethality, although surviving mice had some defects in B-lymphopoiesis and multiple constitutional developmental defects. More recent work presented at the ASH 2011 meeting suggests that conditional deletion of Asxl1 in a hematopoietic-specific context appears to result in morphologic myelodysplasia and the development of anemia and leukopenia with a latency of at least 6 months after deletion, and that Asxl1 loss collaborates with other oncogenic alleles in vivo.74
Beyond epigenetics: spliceosomal mutations in MDS patients
Recent landmark papers by Yoshida et al,76 Papaemmanuil et al,77 and Graubert et al78 presented the discovery of somatic mutations in multiple members of the spliceosome in patients with MDS. From these studies, it was shown that mutations in spliceosomal components in MDS patients appear most commonly in SF3B1, U2AF1, SRSF2, and ZRSR2 (Table 1). In addition, the mutations in each of these genes appear to occur most commonly as heterozygous missense mutations and are rarely cooccurring, suggesting that the mutations may confer a gain-of-enzymatic activity or result in a dominant-negative function.
Despite the mutual exclusivity of mutations in spliceosomal members, mutations in SF3B1 appear to be remarkably enriched in patients with refractory anemia with ringed sideroblasts (RARS) compared with other myeloid malignancies, and have been reported in as many as 75% of these patients.76 Multiple studies have suggested that SF3B1-mutated RARS patients, an already relatively favorable subset of MDS patients, may have improved outcome compared with their wild-type counterparts.63,79 In contrast, mutations in SRSF2, U2AF1, and possibly ZRSR2 appear to be more commonly associated with other subsets of de novo MDS, occurring in 1.5%-11.6%, 8%-12%, and 1.4%-8.0% of MDS patients, respectively.63,80,81 Three independent studies found that mutations in SRSF2 appeared to be associated with higher-risk MDS subtypes,63,80,81 and in 2 of the 3 studies, SRSF2 mutations were important predictors of worsened OS in MDS, possibly due to higher rate of leukemic transformation.63,80
The functional consequences of these mutations are still being elucidated; however, studies to date seem to indicate that the effect of these mutations may not be universal across all spliceosome members. Studies by Graubert et al on the effect of U2AF1 mutations on splicing using a luciferase reporter construct in a transient transfection assay suggested that these alterations may actually enhance splicing of pre-mRNA.78 Conversely, experiments performed by Yoshida et al on the effects of wild-type versus mutant U2A35 expression actually suggested that the mutant form of U2A35 resulted in an inhibition of RNA splicing.76 Using gene-expression profiling of HeLa cells transfected by wild-type and mutant U2A35, those investigators found that mutant forms of U2A35 led to the induction of members of the nonsense-mediated decay pathway and increased generation of unspliced transcripts. To determine the effect of SF3B1 mutations on transcription in MDS patients, Papaemmanuil et al performed gene-expression profiling in 56 MDS patients (12 of whom had SF3B1 mutations) and exon-specific expression profiling in 12 MDS patients (6 of whom had SF3B1 mutations). This analysis revealed down-regulation of gene sets in key pathways determining mitochondrial function; however, very few genes revealed differences in exon usage between patients with and without SF3B1 mutations and none had clear relevance to MDS.77 Given the potential confounding effect of multiple coexisting mutations in primary patient samples, Yoshida et al performed functional analyses of specific spliceosomal mutations in an isogenic context. In vitro analyses of cells transduced with wild-type and mutant forms of U2AF35 by this group revealed that cells induced to express mutant forms of U2AF35 had a growth disadvantage, with a marked increase in cells with G2/M arrest and enhanced apoptosis. Likewise, overexpression of mutant forms of U2AF35 in hematopoietic stem/progenitor cells from mice followed by competitive-reconstitution assays also revealed a competitive disadvantage of the U2AF35 mutant cells at 6 weeks compared with wild-type controls.76 Further work using stable expression of wild-type and mutant forms of the spliceosomal proteins in correct stoichiometric relationships using the endogenous promoter of these proteins will hopefully clarify the effect of splicing alterations on pre-mRNA splicing and HSC function. Determining the functional implication of spliceosomal gene mutations on mRNA processing may have important therapeutic potential, because mutations that confer a gain in function may be more readily targetable therapeutically. Compounds that specifically target the SF3A/B subunits of U2 snRNP to result in nuclear export of intron-bearing precursors exist and could be studied further to determine whether they interfere with the aberrant splicing due to recurrent mutations.82,83
Summary and conclusions
An incredible amount of progress has been made in the past few years toward the elucidation of the molecular mechanisms behind the development of MDS. Novel mutations have been described that target the epigenetic machinery and may explain the profoundly aberrant epigenetic profiles that had been observed previously in this disease. Likewise, mutations targeting the mRNA splicing pathway have also been described, some of which may be responsible for specific forms of MDS such as RARS. The precise impact that these mutations have on clinical outcome has not been fully established for all of these mutations and, given the relatively low frequency of some of them, larger clinical trials will be necessary to more accurately assess this impact. At the same time, the identification of a series of recurrent molecular genetic alterations in MDS patients with prognostic importance and diagnostic specificity suggests that implementing comprehensive molecular analysis of these mutations into clinical practice may be useful. The current limitations preventing implementation of sequencing of these genes into real-time clinical practice include the high cost, slow turnaround time, and lack of clinical validation. However, efforts to limit the sequencing to panels of target genes may improve all of these limitations. Array-based sequencing using hybrid capture technology may be time-saving compared with other sequencing approaches and is cost-effective compared with PCR-based methods.
Whereas initial functional studies in vitro and in vivo have begun to establish a causative role between some of these mutations (eg, ASXL1 and TET2 mutations) and the development of MDS, the exact mechanism by which these genetic alterations lead to the development of this disease will require more comprehensive functional studies. Moreover, given that many of these mutations are common among patients with MDS in addition to other myeloid malignancies such as AML and MPNs, more work will be needed to understand their unique contribution to the development of dysplasia and ineffective hematopoiesis. A different pattern of mutational cooccurrences between MDS patients and patients with other myeloid malignancies, the precise timing and order of acquisition of these mutations, and/or the contribution of stem cell–extrinsic factors unique to MDS patients may be responsible for the phenotype associated with the diagnosis of MDS. Biochemical and epigenomic studies to uncover the direct effect of alterations in individual epigenetic modifiers on DNA methylation and/or histone posttranslational modifications and to identify specific genetic targets that may be responsible for hematopoietic transformation will be needed.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Maria E. Figueroa MD, Department of Pathology, University of Michigan, 109 Zina Pitcher Place, 2019 BSRB, SPC 2200, Ann Arbor, MI 48176; Phone: 734-763-1865; Fax: 764-763-2162; e-mail: marfigue@med.umich.edu.