Abstract
A 62-year-old man with a history of diabetes and hypertension is referred to your hematology clinic for an incidental discovery of anemia. He does not have any constitutional symptoms and previous blood counts have been within the normal range. He has hepatosplenomegaly with a palpable spleen of 6 cm below the left costal margin and a liver size of 2.5 cm below the right costal margin. Laboratory evaluation shows a WBC count of 12.8 K/μL, hemoglobin of 11.0 g/dL, and platelets of 202 K/μL, with a mean corpuscular volume of 85.7, 72% neutrophils, 13% lymphocytes, 4% monocytes, 5% eosinophils, 1% basophils, 1% promyelocytes, 4% myelocytes, and lactate dehydrogenase of 447 U/L (upper limit of normal is < 340 U/L). Peripheral blood smear shows 2+ teardrop-shaped RBCs, large hypogranular platelets, and rare nucleated RBCs. Bone marrow (BM) biopsy exhibits a hypercellular BM with atypical megakaryocytes and increased reticulin fibrosis (MF-1). BCR-ABL gene rearrangement by FISH was negative and JAK2 V617F mutation was 95% positive. He was diagnosed with primary myelofibrosis considered low risk (risk score of 0) by the International Prognostic Scoring System.1 Because he is low risk and asymptomatic, he does not need treatment at this time.2 However, he has read about the possible clinical benefits of IFN-α and its potential reduction of BM fibrosis and wonders whether this would be an appropriate treatment.
Introduction
Primary myelofibrosis (PMF) is a Philadelphia chromosome–negative myeloproliferative neoplasm that presents clinically with abnormal blood cell counts (anemia being the most common feature, along with thrombocytosis/thrombocytopenia, and leukocytosis/leukopenia) and BM failure related to ineffective hematopoiesis and progressive BM fibrosis; splenomegaly due to extramedullary hematopoiesis; and debilitating symptoms related to the combined effects of massive splenomegaly and elevated proinflammatory cytokines. Median survival from the time of PMF diagnosis depends on specific prognostic factors and can range from 135-27 months for low- to high-risk disease, respectively, based on the International Prognostic Scoring System (IPSS).1 Overall survival and progression to acute myeloid leukemia can also be evaluated during the course of the disease by using 2 further refined dynamic prognostic scoring systems that also incorporate acquisition of anemia during follow-up (DIPSS),3 and karyotype, RBC transfusion dependence, and platelet count (DIPSS-Plus) as additional clinicopathologic variables.4
Historically, PMF has been difficult to treat. Only myeloablative hematopoietic stem cell transplantation has curative potential, but also carries the risk of treatment-related morbidity and mortality and is suitable for only a minority of patients. Other therapies, such as hydroxyurea and immunomodulatory agents such as thalidomide, lenalidomide (with or without corticosteroids), and, more recently, pomalidomide, are palliative. Ruxolitinib, the first-in-class JAK inhibitor recently approved by the US Food and Drug Administration (FDA) for PMF therapy, and other JAK inhibitors currently being tested in clinical trials have been very useful for alleviating disease-related constitutional symptoms and painful splenomegaly, but some of these agents have limiting toxicities such as anemia and thrombocytopenia. In addition, although some preliminary data obtained in a few patients in early phase trials seemed promising, no significant decrease in JAK2 mutant allele burden or in BM fibrosis was consistently reported with JAK inhibitors. Due to its known biologic effects on clonal erythroblastic and megakaryocytic differentiation in vitro, hematopoietic stem cell cycling and proliferation, and antiangiogenic properties,5 recombinant IFN-α initially appeared to be a good candidate as a disease-modifying treatment, but early trials did not yield encouraging responses, mainly due to a high incidence of side effects leading to early treatment discontinuation. More recently, studies with pegylated IFN-α-2a have shown more promising results, particularly with “early” stage PMF with low-grade (grade 1-2) fibrosis. To examine the effects of IFN-α (standard and pegylated), we performed a comprehensive literature review of clinical studies using IFN-α in all stages of PMF.
Methods
A literature search was performed by combining the MeSH terms “primary myelofibrosis” (no restrictions, 5809 hits) and “interferon-alpha” (no restrictions, 58 190 hits) between 1966 and 2012, which produced 107 citations. We excluded 28 reviews, 25 non-PMF–related citations referring to secondary myelofibrosis, 22 case reports, 11 nonclinical reports, 4 non-English reports without available abstracts, 3 studies in which IFN-α was combined with other treatments and thus was not separately analyzable, and 1 symposium report. Two additional trials from Hasselbalch et al and Radin et al were included that did not use “primary myelofibrosis ” or “interferon alpha” as a MeSH term.6,7
Results
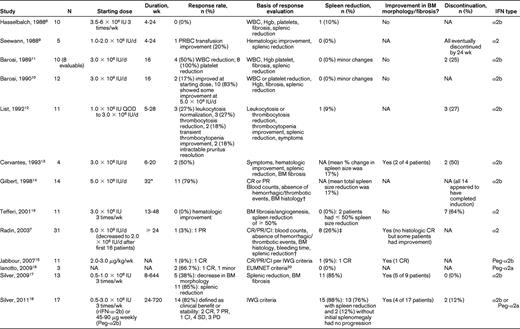
Table 1 summarizes the 13 relevant clinical studies found in our search. These studies overall had a relatively small number of patients (ranging from 3-31), particularly when PMF patients were evaluated as part of a larger cohort of myeloproliferative neoplasms. Nine of the 13 studies were reported before publication of the International Working Group (IWG) consensus response criteria8 for myelofibrosis in 2006 and thus did not use a consistent definition of treatment response.6,7,9–14 Using individual response criteria, overall response rates ranged from 0%-79%, with the highest response in the Gilbert et al study, in which 11 of 14 patients achieved a complete or partial response by the investigators' own definition.14 By physical examination, spleen reductions of more than 1 cm occurred in 0%-26% of patients, but this did not necessarily mean a ≥ 50% spleen reduction was achieved. Two of 6 studies with available BM results showed a reduction in BM fibrosis or improvement in BM morphology.13
Four studies were published after 2006, with cohorts ranging from 3-17 patients, including 3 studies using pegylated IFN-α2a or IFN-α2b.15–19 IWG or European Myelofibrosis Network (EUMNET)20 criteria (2005) were used more consistently in these studies. Overall response rates ranged from 9%-85%, with at least 4 patients reported to have achieved a complete response. Palpable spleen sizes decreased in 0%-76% of patients, with 12% of patients without initial splenomegaly showing no splenic progression, and 10 patients had some improvement in BM fibrosis and morphology.15–16,18 One trial and its follow-up by Silver et al focused on early-stage PMF and reported that more than 80% of the 17 patients included derived clinical benefit or stability per IWG criteria, with improvement in BM morphology in 4 patients. In this trial, 76% of patients achieved spleen reduction, with 9 of 15 (60%) patients with initial splenomegaly achieving complete resolution of splenomegaly (median, 5.0 cm; range, 1.0-16.0 cm) and 15 of 17 (88%) patients having either a decrease in spleen size or no progression if splenomegaly was not initially present.18 The investigators in that study concluded that the use of low-dose IFN-α in morphologically “early” phase PMF resulted in BM improvement or fibrosis reversion, reduction in splenomegaly, and disease stabilization in a clinically significant proportion of patients and had acceptable toxicity.18
Conclusion
A comprehensive review of the effects of IFN-α in PMF is complicated by the heterogeneity of the clinical studies available, with substantial variability in the definitions of treatment response, use of different IFN formulations, different dosing regimens and durations of treatment, incomplete reporting of baseline patient characteristics, and difficulty assessing response rates specifically attributable to “early” versus “advanced” PMF patients, which may have confounded any promising trends in early-stage PMF treatment. As suggested by Silver et al, however, there may be a role for IFN-α in early-stage patients with low-grade BM fibrosis with residual hematopoiesis18 ; patients with more advanced stages of PMF should be considered for other therapies—including hematopoietic stem cell transplantation when feasible, JAK inhibitors, or immunomodulatory drugs—or should be recommended for clinical trials using new investigational agents or combination therapies. The use of pegylated forms of IFN-α may also improve tolerance and allow longer durations of therapy, which is crucial for achieving meaningful response in PMF.
We conclude that although there are limitations in the consistency of the interpretations of patient responses, the overall experience of IFN-α therapy in all-comers of PMF have generally been disappointing. However, several investigators have reported a significant benefit of IFN-α in early-phase PMF. Based on the evidence available as a whole, which consists mainly of observational studies, we recommend (grade 2C) that IFN-α may have a beneficial role in PMF patients with the following profile: (1) absence of massive splenomegaly (ie, < 10 cm), (2) absence of marked leukopenia or thrombocytopenia, and (3) low-grade (grade 1-2) BM fibrosis. In such patients, IFN-α therapy may be considered and may result in significant benefit when used at low doses (to improve tolerance) and for a sufficient duration (> 12 months).16,18 Large, randomized controlled trials are warranted to confirm the findings derived from the published small trials presented in this review, and many other strategies and new agents are currently being evaluated.
Disclosures
Conflict-of-interest disclosure: J.-J.K. is on the board of directors or an advisory committee for Shire, Novartis, and Incyte and has received research funding from Novartis and Celgene. H.N. declares no competing financial interests. Off-label drug use: IFN-α is not registered for the therapy of PMF.
Correspondence
Huong (Marie) Nguyen, Stanford University School of Medicine, 269 Campus Dr, CCSR 1155, Stanford, CA 94305; Phone: 650-723-7078; Fax: 650-736-0974; e-mail: nguyenht@stanford.edu or Jean-Jacques Kiladjian, Centre d'Investigations Cliniques, Hôpital Saint-Louis, 1 avenue Claude Vellefaux, 75010 Paris, France; Phone: 33-1-42-49-94-94; Fax: 33-1-42-49-93-97; e-mail: jean-jacques.kiladjian@sls.aphp.fr.