Abstract
B- and T-cell subtypes of prolymphocytic leukemia (PLL) are rare, aggressive lymphoid malignancies with characteristic morphologic, immunophenotypic, cytogenetic, and molecular features. Prognosis for these patients remains poor, with short survival times and no curative therapy. The advent of mAbs has improved treatment options. In B-PLL, rituximab-based combination chemoimmunotherapy is effective in fitter patients. TP53 abnormalities are common and, as for chronic lymphocytic leukemia, these patients should generally be managed using an alemtuzumab-based therapy. Currently, the best treatment for T-PLL is IV alemtuzumab, which has resulted in very high response rates of more than 90% when given as frontline treatment and a significant improvement in survival. Consolidation of remissions with autologous or allogeneic stem cell transplantation further prolongs survival times, and the latter may offer potential cure. The role of allogeneic transplantation with nonmyeloablative conditioning needs to be explored further in both T- and B-PLL to broaden the patient eligibility for what may be a curative treatment.
Introduction
A 55-year-old man presents with a 2-month history of sweats and abdominal discomfort. He has massive splenomegaly, a WBC count of 250 × 109/L, and a peripheral blood film showing a homogeneous population of medium-sized lymphoid cells with a prominent nucleolus and basophilic cytoplasm. Twelve months earlier, he had been noted to have a mild lymphocytosis of 20 × 109/L, which was unchanged when rechecked 6 months later, and he was thought to have a stable early-stage chronic lymphocytic leukemia (CLL). This patient had features compatible with a diagnosis of prolymphocytic leukemia (PLL), a disorder first described in the 1970s.1 At that time, immunophenotyping to distinguish B-PLL from T-PLL and the extensive range of tests, both immunologic and genetic, required to discriminate between a variety of B- and T-cell leukemias would not have been available. In this patient, the same clinical presentation and peripheral blood morphology could represent either the B- or the T-cell subtype. However, in terms of biology and management, the similarity stops there. Some 40 years later, we have a better understanding of the biologic diversity within the mature lymphoid malignancies and the laboratory tools are now available to make a precise diagnosis.2 Sadly, our ability to treat PLL successfully has not made such a rapid advance. Neither B-PLL nor T-PLL respond well to conventional alkylator-based therapies, but the advent of mAbs, used alone or in combination regimens, has improved the outlook for these patients. The strategies are very different for the 2 subtypes. After a brief review of the presentation and diagnosis of PLL, the main focus herein will be on the management of these leukemias with mAb-based therapy.
Clinical presentation and laboratory diagnosis of PLL
B-PLL and T-PLL are extremely rare, accounting for only approximately 1%-2% of lymphocytic leukemias. Presentation is usually in the sixth decade or later and occurs more frequently in males (Table 1).3,4 As outlined in the example above, there is considerable overlap in the clinical features between the B- and T-cell subtypes of PLL. Both characteristically have rapidly increasing lymphocyte counts and splenomegaly. Low-volume lymphadenopathy, skin rashes, peripheral edema, and pleuroperitoneal effusions are seen relatively frequently in T-PLL but not in B-PLL. CNS involvement has been described in both, but is rare.
A minority of patients may be asymptomatic at diagnosis and this “indolent ” phase can persist for a variable length of time, although rarely for more than 1-2 years. However, progression can be very rapid when it occurs and patients should therefore be monitored closely.
Diagnosis requires a systematic approach and careful integration of the results of morphology (particularly of the peripheral blood) with specialist diagnostic tests including immunophenotyping, cytogenetics, and molecular genetics.
For a diagnosis of B-PLL, prolymphocytes should account for more than 55% of peripheral blood lymphoid cells, and the proportion usually exceeds 90%. Prolymphocytes are medium-sized cells (twice the size of a CLL lymphocyte) with a high nuclear-cytoplasmic ratio, a single prominent nucleolus, and basophilic agranular cytoplasm. No cytoplasmic hairy projections or “villi ” are seen in B-PLL in contrast to the hairy cell leukemia variant (HCL-V) and splenic marginal zone lymphoma (SMZL). In B-PLL and in 50% of T-PLL cases, the cells have a round to oval nucleus. In the remainder of T-PLL cases, the nuclei are irregular, often with convolutions, although these are less pronounced than those seen in Sézary syndrome (SS) or adult T-cell leukemia/lymphoma (ATLL) cells. In typical T-prolymphocytes, the cytoplasm is more intensely basophilic and has an irregular outline with “blebs.” Two variants, small-cell (previously known as T-CLL, a term no longer used) and cerebriform, are seen in approximately 20% of cases.2 Both of these variants have immunophenotypic, cytogenetic and clinical features which are similar to typical T-PLL. In B-PLL, BM, lymph node, and spleen histology may all be important in confirming the diagnosis, whereas in T-PLL, these are rarely needed.
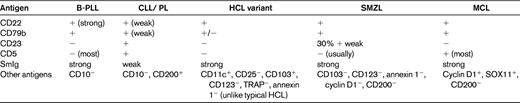
B-PLL can readily be distinguished from its T-cell counterpart by immunologic markers. In B-PLL, the monoclonal B-cell proliferation is confirmed by establishing light-chain restriction and the B cells are further characterized by use of a panel of immunophenotypic reagents (Table 2). This will rule out the presence of typical CLL (CD5+, CD23+, weak surface Ig, and CD79b)5 or CLL with an increase in prolymphocytes (CLL/PL), which has the same phenotype. In contrast, in B-PLL, there is strong Ig and CD79b expression and most cases are CD23− and CD5−. T-prolymphocytes have a postthymic (TdT−, CD1a−) T-cell phenotype (CD5+, CD2+, CD7+) with variable expression of CD4 and CD8.3 Not all cases will express membrane CD3, although this is invariably present in the cytoplasm and CD7 expression is strong, in contrast to other mature T-cell leukemias, in which this marker is often weakly positive or negative. CD25, CD38, and class II HLA-DR may be variably expressed, but markers identifying cytotoxic T cells such as TIA-1 are negative, even in cases with a CD8+ phenotype.
The most common cytogenetic abnormality seen in B-PLL is del 17p involving loss of TP53.6 t(11,14), the hallmark translocation for mantle cell lymphoma (MCL), is not seen.7 Rarely there are translocations involving C-MYC.8 The majority of T-PLL cases will have complex karyotypes, typically with abnormalities involving chromosome 14, and frequently also chromosomes 8 and 11.9 These changes result in the activation of oncogenes (TCL-1, MTCP-1, and ATM) involved in the pathogenesis of this disorder.10–12
Discrimination of B-PLL and T-PLL from other mature B- and T-cell malignancies with a leukemic presentation can be challenging. Indeed, the existence of B-PLL as a separate entity is disputed by some. Mature B-cell leukemias with a similar presentation include CLL (particularly CLL/PL), MCL, SMZL, and HCL-V. The percentage of prolymphocytes (> 55%), the immunophenotype (Table 2), and the absence of proliferation centers in the BM biopsy help to separate B-PLL from CLL/PL. True de novo B-PLL does not arise on a background of known CLL. The demonstration of t(11;14) and positive expression of cyclin D1 and/or SOX 11 is able to identify cases of MCL with a leukemic presentation.13 Precise diagnosis of B-PLL can be problematic because of its similarity to both SMZL and HCL-V in terms of its clinical presentation (older age, splenomegaly, and lymphocytosis), the overlap in morphologic features (cytoplasmic basophilia, nucleolus in HCL-V, and the loss of the fine “hairy ” projections in poorly prepared SMZL and HCL-V films), together with a lack of distinct immunophenotypic or cytogenetic markers. Spleen histology, when available, can be helpful.14 The presence of B symptoms, a very high WBC count (> 100 × 109/L), and aggressive clinical course are much more characteristic of B-PLL than of SMZL or HCL-V. The distinction is important therapeutically, because many cases of HCL-V and SMZL do not require therapy or may benefit from splenectomy or rituximab monotherapy alone, whereas B-PLL is likely to require a combination chemoimmunotherapy approach.
The morphologic and immunophenotypic features of T-PLL help to distinguish it from other postthymic T-cell malignancies, which include: T-cell large granular lymphocytic leukemia, ATLL, SS, and peripheral T-cell lymphoma. In addition, T-PLL is negative for HTLV-1. Hypercalcemia, which is common in ATLL, is rare in T-PLL. Skin histology is distinct between T-PLL and SS, with dermal infiltration, preferentially around the appendages and sparing the epidermis in T-PLL, in contrast to the characteristic epidermotropism and microabscesses seen in SS.
Further characterization of PLL has occurred after the advent of more sophisticated molecular analysis, which may advance our understanding of the biology of these disorders and identify potential new therapeutic targets.15–18
Therapy of PLL
The rarity of PLL has meant that there have been no prospective randomized controlled studies to compare the efficacy of different therapies. Indeed, even nonrandomized studies are mostly retrospective, with small patient numbers accrued over prolonged periods of time. Many of the reports, especially for B-PLL, are of only 1 or 2 patients. There are several problems with this type of data, including the bias in reporting and the difficulty of comparing small studies. Entering patients into clinical trials is therefore to be encouraged.
PLL, either B- or T-cell, responds poorly to conventional alkylator-based therapies. Improvements in outcome have arisen after the introduction of mAb approaches, including the anti-CD20 Ab rituximab in B-PLL and the anti-CD52 Ab alemtuzumab in both B-PLL and T-PLL; neither of these Abs is licensed for these specific treatment indications.
Rituximab in B-PLL
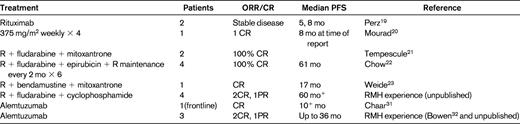
Rituximab is a chimeric anti-CD20 mAb widely used in B-cell malignancies. However, supporting data for any specific therapeutic recommendations in B-PLL are very limited, and it is necessary to draw on the experience gained in other related B-cell disorders. There are case reports documenting the successful treatment of B-PLL with rituximab monotherapy, although the durability of these responses appeared to be short.19,20 Combinations of rituximab with fludarabine or bendamustine together with an anthracycline such as mitoxantrone or epirubicin (FMR, FER, and BMR) have also been reported to have activity in B-PLL21–23 (Table 3). Given the excellent responses seen in CLL and MCL with the combination of fludarabine, cyclophosphamide, and rituximab (FCR), my usual approach to fit patients without TP53 abnormalities is to use this regimen as frontline therapy. In our experience, this has induced durable complete remissions (CR) in 2 of 4 patients lasting for more than 5 years. Because bendamustine plus rituximab has been shown to have efficacy in CLL and other B-cell malignancies, this could also be a very suitable therapy, and may be associated with less hematologic toxicity. I follow the algorithm in Figure 1.

Treatment algorithm for PLL. BR indicates bendamustine + rituximab; NR, no response; RT, radiotherapy; FIC, full-intensity conditioning; RIC, reduced-intensity conditioning.
Treatment algorithm for PLL. BR indicates bendamustine + rituximab; NR, no response; RT, radiotherapy; FIC, full-intensity conditioning; RIC, reduced-intensity conditioning.
New anti-CD20 mAbs (eg, ofatumumab and GA101) have not been evaluated in B-PLL although it is likely that activity will be similar to that seen in CLL. In addition, the new B-cell receptor antagonists targeting molecules such as Brutons Tyrosine Kinase and PI3 Kinase delta, may also have activity in B-PLL, including in those cases with TP53 abnormalities.
Alemtuzumab
Alemtuzumab (campath-1H) is a humanized IgG1 Ab, targeting the CD52 antigen which is highly expressed on normal and malignant T- and B- lymphocytes and monocytes but not on hemopoietic stem cells. In vitro the Ab can induce cell death by Ab- dependent cellular cytotoxicity (ADCC), complement activation and possibly also through direct apoptosis, although which of these mechanisms are operative in vivo is not fully elucidated.
Alemtuzumab in T-PLL
Currently the best treatment for T-PLL is alemtuzumab followed by consolidation with a hematopoietic stem cell transplantation (HSCT) where possible. This approach has led to an extension of the median survival from 7 months in our historic series of more than 70 patients treated with conventional chemotherapy to more than 4 years for those patients receiving alemtuzumab followed by HSCT.24 I follow the algorithm in Figure 1.
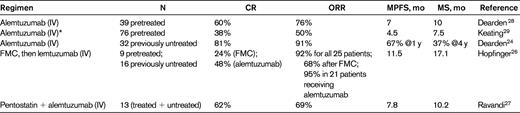
We have now treated a total of 88 T-PLL patients with single agent alemtuzumab at the standard dose of 30mg 3 times a week until maximal response: 45 previously treated patients and 43 who were therapy-naive. Nine of the patients treated frontline were enrolled on a pilot study to evaluate the subcutaneous (SC) route of administration of alemtuzumab.24 This pilot study was terminated early because of the dramatic fall in response rates associated with the change to SC administration. IV alemtuzumab results in overall response rates (ORRs) in excess of 90%, with 81% CRs when given to previously untreated patients with T-PLL (Table 4).24 The ORR decreased to only 33% when the Ab was administered SC. It was possible to rescue a proportion of these patients by switching to IV administration and/or by adding pentostatin, but 2 of 9 patients died on treatment. The likely reason for this poor result is the longer delay in achieving peak Ab levels via the SC route,25 which may be critical in this rapidly progressive leukemia. Alternatively, the poor result may be because SC administration in a previously untreated patient could be sufficiently immunogenic to induce neutralizing Abs. On the basis of these results, I always use IV administration of alemtuzumab in patients with T-PLL.
Skin disease responds very well to alemtuzumab therapy. However, for patients with CNS disease, it is necessary to administer CNS-directed therapy, either triple (methotrexate 12.5 mg, cytarabine 50 mg, and hydrocortisone 12.5 mg) intrathecal or high-dose systemic methotrexate (3 g IV). We do not use routine CNS prophylaxis because this complication is seen in < 10% of patients. Treatment failures are in a minority and are most often seen in patients with extramedullary disease, such as serous effusions and liver involvement. Adding another agent, such as a purine analog, may induce remission in these circumstances.
We have found alemtuzumab to be well tolerated in the T-PLL patient population, with fewer infectious complications than when it is used in patients with relapsed/refractory CLL (approximately 10% in T-PLL vs 40% in CLL in our institution). Hematologic toxicity (apart from profound and prolonged lymphopenia) is minimal. The most serious complications of treatment are usually related to infection, often with atypical organisms and viruses. Careful attention to infection prophylaxis for Pneumocystis jiroveci and herpes viruses, together with regular weekly monitoring for CMV reactivation (by PCR), have minimized serious infections. Infusion reactions are common on initiating IV therapy, but can be readily controlled with the use of premedication and rarely last beyond the first week of treatment. None of our patients has developed tumor lysis.
Therefore, alemtuzumab administered IV as a single agent will induce remissions in the majority of T-PLL patients treated frontline, with minimal toxicity. Remarkably, these responses occur regardless of the apparent bulk of the disease at presentation (ie, high WBC counts, high lactate dehydrogenase levels, and splenomegaly). It is not advisable to implement a debulking strategy using steroids or multiagent regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), because this is usually ineffective, delays starting more effective Ab therapy, and adds to toxicity.
Combination Ab therapy, either given sequentially or concurrently, has also been explored in T-PLL. The German CLL study group (GCLLSG) has conducted a prospective phase 2 trial in 25 patients with previously treated (n = 9) and treatment-naive (n = 16) T-PLL.26 The sequential therapy comprised up to 4 cycles of FMC (fludarabine, mitoxantrone, cyclophosphamide) given every 4 weeks, followed by consolidation with IV alemtuzumab 3 times a week in responding patients 1-3 months after completion of chemotherapy. ORR was 68% after FMC (6 CRs) and 92% after both therapies. Alemtuzumab consolidation in 21 patients increased the ORR to 95% in these patients (80% of all patients) with a doubling of the CR rate (12 CRs). Median overall and progression-free survival were 17 and 12 months, respectively. The MD Anderson Cancer Center has explored the use of alemtuzumab in combination with pentostatin in a range of peripheral T-cell lymphomas, including T-PLL, and found activity (ORR 69%) similar to that seen with alemtuzumab alone.27 These treatment trials are summarized in Table 4.
Despite the very high response rates with alemtuzumab, relapse is inevitable and median survival remains only 20 months. For those patients who have not previously received alemtuzumab, this is the treatment of choice at relapse, and good response rates have been documented in the relapsed/refractory (alemtuzumab naive) patient group.28,29 We have previously reported the results of a study using IV alemtuzumab in 39 previously treated relapsed/ refractory patients, which showed 60% CRs and 16% partial remissions.28 The median disease-free interval after therapy was 7 months (range, 4-45). Longer follow-up of this series shows a median survival of 2 years for those patients achieving a CR and 9 months for those in partial remission. For those patients who have previously received induction therapy with alemtuzumab with a response duration of at least 6 months, retreatment can be successful in achieving a second, or even third, remission (> 50% of patients will respond a second time), but this is usually of much shorter duration. Occasionally, the T-PLL cells lose expression of the CD52 antigen, precluding further use of alemtuzumab, and it is therefore important to retest for this at relapse.30 Maintenance alemtuzumab has not been formally evaluated and is likely to encourage early loss of CD52 expression. For those patients who fail or are unsuitable for alemtuzumab retreatment, a purine analog–based therapy or experimental agent within a clinical trial should be undertaken, but response rates are not high. If the patient is a suitable candidate for an allogeneic HSCT, then it is sometimes possible to induce a remission with an intensive combination regimen and proceed directly to HSCT. With the current treatment options, very few patients will have a successful outcome after relapse. The advent of novel small-molecule inhibitors targeting dysregulated growth and survival pathways in T-PLL may improve the outlook in the future and may even supersede current treatment options.
Alemtuzumab in B-PLL
Alemtuzumab is also important in the therapy of B-PLL patients who have deletions and/or mutations of TP53. As in CLL, this genetic abnormality is associated with inherent chemoresistance and probably explains, at least in part, the poor outcome seen in B-PLL, in which up to 50% of patients harbor this defect. Alemtuzumab is also most active in the blood, BM, and spleen, which are the main sites involved in B-PLL, whereas bulky lymphadenopathy is almost never seen. Alemtuzumab can also be an effective salvage therapy for patients refractory to purine analog–based treatment; there have been some single case reports of this.31,32 We have seen a CR in 2 patients with B-PLL after alemtuzumab. In both patients, a TP53 deletion was detected after failure to respond to FCR. In the first of these, a 48-year-old man, alemtuzumab was followed by a sibling allogeneic HSCT. That patient remains well in continued remission 6 years later. The other patient remained in remission for 36 months after achieving a CR with alemtuzumab. He subsequently relapsed with a high-grade transformation to diffuse large B-cell lymphoma. He obtained a CR after R-CHOP, went on to receive an unrelated reduced intensity allograft, and remains well 6 months later. Some of the new agents currently being evaluated, particularly the BCR antagonists, may have activity in B-PLL, including in those patients with a TP53 abnormality.
HSCT
Although some responses with alemtuzumab are very prolonged (more than 5 years), longer-term follow-up on T-PLL patients treated with alemtuzumab in our series suggests that all patients do eventually relapse.24 Median survival remains only 20 months for those patients achieving a durable CR after alemtuzumab monotherapy, with a very similar outcome in the German trial of FMC followed by alemtuzumab (19 months).26 Our experience suggests that survival may be prolonged by the use of autologous or allogeneic HSCT.
We have recently published our data on 28 T-PLL patients treated on a common protocol with alemtuzumab followed by either autologous (n = 15) or allogeneic (n = 13) HSCT,33 and compared clinical outcomes with 23 retrospectively selected patients who had achieved a CR after alemtuzumab treatment and survived at least 6 months but who had not undergone HSCT (non-HSCT group). Apart from age (median, 64 years in the non-HSCT group vs 58 for the autograft group and 51 for the allograft group), the clinical and disease characteristics of the retrospective cohort were the same as those of the patients undergoing HSCT. Overall survival was similar in the autograft and allograft groups at a median of 48 months, compared with a median survival for the non-HSCT group of 20 months. There was no association between age and survival in either group. Of our series of more than 80 patients treated with alemtuzumab, almost half of those achieving remission have proceeded to either autologous or allogeneic HSCT. The remainder were either unsuitable or unwilling to undergo HSCT. The introduction of reduced-intensity conditioning has minimized the transplantation-related mortality, and it is hoped that after longer follow-up, this will translate into improved survival for the allograft group. We have not seen any failure of engraftment despite the prior use of alemtuzumab, although we usually allow at least 3 months between completing induction treatment and the allogeneic HSCT. Outcome for patients who relapse after HSCT is poor and the role of donor lymphocyte infusion is unclear. There has been a retrospective review of the European Group for Blood and Marrow Transplantation (EBMT) database, with 41 T-PLL patients identified who have undergone an allogeneic HSCT.34 The 3-year progression-free and overall survival were 19% and 21% respectively. The 3 year nonrelapse mortality and relapse rate were each 41%, with the majority of relapses occurring in the first year. The main difference between this group of patients and our own allogeneic HSCT series was the proportion of patients in CR at the time of transplantation (11 of 41 for the EBMT series vs 10 of 13 for our series). In multivariate analysis, factors associated with longer progression-free survival were the use of total body irradiation in the conditioning regimen and a shorter interval between diagnosis and HSCT.
Autologous HSCT also benefits patients, prolonging remissions with much less treatment-related toxicity than allogeneic HSCT, but not resulting in cure. It may, however, be a more suitable option for older, less fit patients or those without a suitable donor. Therefore, we would offer an allogeneic HSCT to T-PLL patients who achieve a remission after induction therapy, are physically fit, and have a suitable donor. In our series, patients up to and including 70 years of age have had successful allografts. Those patients unwilling to undertake allograft or who do not have a donor would be offered an autograft.
In B-PLL, published experience of HSCT is confined to a few successful case reports and one registry study.35,36 The latter was a retrospective review of the Center for International Blood and Marrow Transplant Research (CIBMTR) database from 1995-2005, which identified 11 patients with B-PLL (median age, 54 years) who had undergone an allogeneic HSCT. With relatively short follow-up, the median PFS was only 3.5 months, with less than one-third of patients alive and disease-free at 1 year.36 Given that B-PLL generally affects an older patient population, fewer will be suitable candidates for allogeneic HSCT, even with the extended eligibility associated with reduced-intensity conditioning regimens. However, we would use the same criteria for selection as for CLL, namely the presence of a TP53 deletion and/or mutation or failure to achieve a durable remission after chemoimmunotherapy.37
It is possible that HSCT may provide benefit for selected patients with PLL, with some achieving long-term survival (> 5 years).
Summary
B-PLL and T-PLL are 2 rare, clinically aggressive but distinct disease entities with characteristic morphologic, immunophenotypic, and molecular features. Careful diagnostic evaluation is needed to discriminate these disorders from other mature B- and T-cell leukemias that may have a similar presentation. Therapeutic options have improved with the use of mAb therapy. Rituximab-based chemoimmunotherapy combinations should be considered as frontline therapy for B-PLL, with alemtuzumab used for those presenting with abnormalities of TP53. In T-PLL, alemtuzumab delivered IV now results in the majority of patients achieving durable remissions and prolonged survival. However, Ab therapy is not curative and consolidation with HSCT in the first remission should be considered in suitable patients. In the future, novel agents such as new mAbs and BCR antagonists in B-PLL and small-molecule inhibitors in T-PLL may alter the therapeutic paradigm and improve survival for patients with these rare leukemias.
Disclosures
Conflict-of-interest disclosure: The author is on the board of directors or an advisory committee for Roche and Genzyme and has received honoraria from Roche. Off-label drug use: alemtuzumab for the treatment of T-PLL and TP53-deleted B-PLL.
Correspondence
Dr Claire Dearden, Department of Haemato-Oncology, The Royal Marsden NHS Foundation Trust, Downs Road, Sutton, Surrey SM2 5PT, United Kingdom; Phone: 208-661-3116; Fax: 208-642-9634; e-mail: Claire.Dearden@rmh.nhs.uk.