Abstract
Chronic lymphocytic leukemia (CLL) is a malignancy of mature B cells that depend on host factors in the tissue microenvironment for survival and proliferation. In vitro, CLL cells rapidly undergo apoptosis unless microenvironmental factors are provided that support their survival. Signaling pathways activated in the microenvironment in vivo include the B-cell receptor (BCR) and NF-κB pathways. Thus, CLL is a disease “addicted to the host” and is dependent on pathways that promote normal B-cell development, expansion, and survival; this is particularly true in the case of the BCR signaling cascade. Small-molecule inhibitors of kinases that are essential for BCR signal transduction abrogate the stimulating effects of the microenvironment on CLL cells. The orally administered tyrosine kinase inhibitors fostamatinib and ibrutinib and the phosphatidylinositol 3-kinase inhibitor GS-1101 have induced impressive responses in relapsed and refractory CLL patients, mostly with moderate side effects. Reductions in lymphadenopathy and splenomegaly are seen within weeks and are frequently accompanied by a transient rise in absolute lymphocyte count that is asymptomatic and probably the result of changes in CLL cell trafficking. This review discusses the biologic basis for kinase inhibitors as targeted therapy of CLL and summarizes the exciting early clinical experience with these agents.
Introduction
Oncogenic mutations in kinases have been identified in multiple cancers often leading to successful targeted therapy with kinase inhibitors. The paradigm in hematologic malignancies has been the discovery of the BCR-ABL fusion kinase in chronic myeloid leukemia and its successful targeting by tyrosine kinase inhibitors that changed the natural history of the disease. In contrast, possible disease-relevant mutations in kinases have been a rare finding in chronic lymphocytic leukemia (CLL). The most commonly mutated kinase in CLL is BRAF, with < 2% of patients affected.1 However, kinase inhibitors that target signaling pathways that are essential for B-cell development, in particular, those targeting the B-cell receptor (BCR) have induced striking clinical responses. Here I briefly review these signaling pathways and discuss the ongoing clinical development of kinase inhibitors for the targeted therapy of CLL.
Signaling pathways and their kinases in the pathogenesis of CLL
Biology of CLL
CLL is a malignancy of mature B cells involving blood, bone marrow, and lymphoid tissues.2 CLL is the most common leukemia in Western countries and currently is most often diagnosed from an incidental blood count showing lymphocytosis. The median survival with early-stage disease is 10.7 years, but the clinical course is heterogeneous.3 Two major CLL subtypes are distinguished by the presence or absence of somatic mutations in the immunoglobulin heavy chain variable region gene (IGHV), which encodes part of the antigen-binding domain of the BCR. Patients whose CLL cells express an unmutated IGHV gene (U-CLL) have a more rapidly progressive clinical course than patients whose CLL cells express a mutated IGHV gene (M-CLL). ZAP70, a nonreceptor tyrosine kinase essential for T-cell receptor signal transduction, is expressed in most cases of U-CLL and less frequently in M-CLL. ZAP70 expression correlates with more rapid disease progression in both subtypes defined by IGHV gene mutation status.4
The role of the microenvironment in CLL pathogenesis
CLL cells in the blood are resting cells with a gene expression profile similar to memory B cells.2 However, CLL cells in the lymph node and bone marrow show characteristics of activated B cells and demonstrate increased proliferation.5 In the tissue sites, CLL proliferation is often highest in anatomic structures labeled as “proliferation centers” where CLL cells can interact with other cells, in particular T cells and stromal cells.6 Thus, the biology of CLL cells in vivo depends on their anatomic location and is influenced by extrinsic signals from the tissue-microenvironment. In vitro, CLL cells undergo apoptosis unless appropriate microenvironmental factors are provided. This dependence of CLL cells on pathways that also promote normal B-cell development, expansion, and survival,5–9 indicates that this tumor is “addicted to the host,” constituting an example of a novel concept termed “non-oncogene addiction.”10
The term “microenvironment” collectively describes cellular, structural, and soluble components of the anatomic compartment in which the CLL cells reside.7 In vitro, different types of stromal cells and monocyte-derived cells, designated “nurse-like cells,” promote CLL cell survival.7,9,11 In addition, T cells were shown to be required for CLL cell proliferation in vivo using a xenograft mouse model.12 Extensive in vitro studies have identified many factors that enhance CLL cell survival and promote limited proliferation. These include the BCR, Toll-like receptors (TLR), cytokines, chemokines, CD40, BAFF, integrins, and components of the extracellular matrix.11,13–19 Many of these extrinsic factors activate similar intracellular signaling pathways, most prominently the PI3K/AKT/mTOR, NF-κB, and MAPK pathways as well as the kinases SYK and BTK. It is therefore difficult to estimate to what degree any single factor or pathway may be necessary or sufficient for CLL pathogenesis. Nonetheless, the BCR is increasingly emerging as a pivotal pathway.
BCR signaling in CLL pathogenesis
The BCR consists of a surface transmembrane immunoglobulin (Ig) receptor associated with the Ig α (Igα, CD79A) and Ig β (Igβ, CD79B) chains.20 Two types of signals can emanate from the BCR: a “tonic” survival signal and an antigen-induced activation signal. Expression of a functional BCR is necessary for B-cell development and the survival of all mature B cells. This “tonic” survival signal is independent of antigen and is mediated by PI3Kα and PI3Kδ (Figure 1A). In contrast, antigen-induced BCR signaling activates several tyrosine kinases in addition to PI3Kδ to promote cell growth, proliferation, maturation, and survival. Of the surface Ig isotypes, IgM is of particular importance for antigen-dependent signaling and is the isotype expressed by most mature B-cell malignancies.21 On antigen binding to the BCR, the tyrosine kinases LYN and SYK initiate a signal transduction cascade that involves several additional kinases, adapter molecules, and second messengers (Figure 1B). Genetic mouse models have identified the kinases SYK, BTK, PI3Kδ, and the phospholipase PLCγ2 as essential for BCR signaling.

Upstream events in BCR signaling. (A) BCR signaling in the absence of antigen binding provides a tonic survival signal dependent on PI3K. In this model, the Ras GTPAse TC21 binds to nonphosphorylated tyrosine motifs (black boxes) in Igα and Igβ and activates PI3K-dependent survival signals. PI3Kα and PI3Kδ assume redundant functions in this pathway. (B) BCR signaling in response to antigen binding induces LYN- and SYK-dependent phosphorylation (phosphorylation denoted by “P” in orange circle) of tyrosine motifs (red boxes) on CD79A and CD79B. A number of protein kinases (red symbols) and the lipid kinase PI3Kδ (blue symbol) transmit survival, cell growth, and proliferation signals and regulate cell migration. The transcription factors NF-κB and NFAT are important regulators of BCR-induced gene expression changes. Small-molecule inhibitors of select kinases in the BCR pathway that have demonstrated significant clinical activity are indicated.
Upstream events in BCR signaling. (A) BCR signaling in the absence of antigen binding provides a tonic survival signal dependent on PI3K. In this model, the Ras GTPAse TC21 binds to nonphosphorylated tyrosine motifs (black boxes) in Igα and Igβ and activates PI3K-dependent survival signals. PI3Kα and PI3Kδ assume redundant functions in this pathway. (B) BCR signaling in response to antigen binding induces LYN- and SYK-dependent phosphorylation (phosphorylation denoted by “P” in orange circle) of tyrosine motifs (red boxes) on CD79A and CD79B. A number of protein kinases (red symbols) and the lipid kinase PI3Kδ (blue symbol) transmit survival, cell growth, and proliferation signals and regulate cell migration. The transcription factors NF-κB and NFAT are important regulators of BCR-induced gene expression changes. Small-molecule inhibitors of select kinases in the BCR pathway that have demonstrated significant clinical activity are indicated.
A role for antigenic stimulation in the pathogenesis of CLL is inferred from the observations that CLL cells use a restricted repertoire of IGHV genes22,23 and that some cases express virtually identical BCRs, so-called “stereotyped BCRs,” that recognize shared antigens.24,25 These antigens remain incompletely defined but in many cases may be autoantigens expressed by dying cells.26 Thus, genetic evidence indicates that antigenic signaling plays a part at some point in the ontogeny of CLL. However, the role of continued antigenic signaling in established disease and the relationship of antigenic signaling to disease progression and outcome is an important area of ongoing investigation. These issues have been addressed in a comparative analysis of purified CLL cells isolated concomitantly from the blood, bone marrow, and lymph node of the same patient.5 Major findings include that CLL cells in the lymph node exhibited increased levels of activated SYK and expressed genes indicative of BCR activation and tumor proliferation. Furthermore, both BCR signaling and tumor proliferation were more active in U-CLL, suggesting that the more rapid disease progression of this subtype is the result of increased BCR activation in vivo. Notably, BCR signaling and subsequent downstream activation of the NF-κB and NFAT pathways most strongly accounted for the genes up-regulated in CLL cells in the lymph node, indicating that activation of the BCR is one of the most prominent effects of the tissue microenvironment on CLL biology in vivo.5 Thus, active antigenic signaling continues throughout the disease course of CLL. This conclusion is also supported by the observations of reversible down-modulation of surface IgM expression on CLL cells and the anergic state of some CLL cells, which is probably the result of chronic antigen stimulation in vivo.27,28
The most compelling experimental evidence that the BCR is essential for proliferation and survival of malignant B cells comes from studies in the activated B cell–like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL), which displays “chronic active BCR signaling” resulting in constitutive activation of the NF-κB and PI3K pathways.29 These lymphomas die after knockdown of the BCR components IgM, CD79A, and CD79B and of kinases that transmit the BCR signal, including SYK, PI3Kδ, BTK, and PKCβ.21,29 In CLL, the evidence is more circumstantial. BCR activation in vitro undoubtedly enhances CLL survival and to some degree promotes proliferation.13,14,30–32 Furthermore, a strong response to BCR activation in vitro correlates clinically with more aggressive disease.5,33,34 In addition, ZAP70 expression increases the response of CLL cells to BCR activation in vitro, providing a mechanistic rationale for why ZAP70 expressing CLL progresses more rapidly.34,35 Consistent with these in vitro data, the degree of BCR activation in CLL cells in vivo correlates with increased tumor proliferation and shorter time to progression.5 Together, these findings implicate antigen-dependent BCR activation as a pivotal pathway in the pathogenesis and progression of CLL.
Targeting kinases for the treatment of CLL
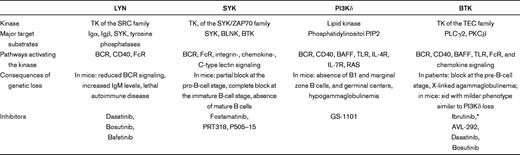
Several kinases in the BCR pathway can be targeted by small-molecule inhibitors. Starting with the most upstream kinase, these are LYN, SYK, PI3K, and BTK (Figure 1B), and they are discussed in that order.
Targeting LYN
The SRC family nonreceptor tyrosine kinase LYN initiates BCR signaling by phosphorylating activation motifs on the Igα and Igβ chains that recruit further components of the signaling pathway. LYN not only phosphorylates (and thereby activates) SYK, but it also activates phosphatases that in turn inhibit signal transduction through the BCR.36 LYN thus controls both activation as well as termination of BCR signaling. Mice deficient in LYN have reduced numbers of B cells. These B cells are less responsive to acute BCR activation but spontaneously produce large amounts of self-reactive IgM, leading to a fatal lupus-like autoimmune disease.37 These data establish that an essential function of LYN is to down-regulate BCR activation and limit the expansion of autoreactive B cells. This is further supported by the discovery that CD79B mutations in ABC-DLBCL reduce LYN kinase activity and promote “chronic active BCR” signaling, resulting in constitutive NF-κB activation.29
LYN inhibitors
Dasatinib is an oral multikinase inhibitor targeting SRC and ABL kinases that is approved for use in imatinib-resistant chronic myeloid leukemia. More recently, it has been shown that dasatinib not only inhibits LYN but also BTK at low nanomolar concentrations.38 In vitro, dasatinib induces variable degrees of apoptosis in CLL cells with no correlation between response and inhibition of LYN phosphorylation. However, induction of apoptosis in vitro was inversely correlated with drug-induced inhibition of SYK phosphorylation. Whereas dasatinib inhibited BCR signaling, stromal cell contact and CD40 stimulation antagonized its proapoptotic effect.39
Clinical experience
A phase 2 study of 140 mg dasatinib once daily that enrolled 15 patients with relapsed/refractory CLL reported an overall response rate (OR) of 20% with a progression-free survival (PFS) of 7.5 months.40 In addition, 5 patients exhibited a > 50% reduction in lymphadenopathy. Myelosuppression was the primary toxicity with grade 4 neutropenia and thrombocytopenia occurring in 40% and 13% of patients, respectively.
Targeting SYK
SYK and ZAP70 are cytoplasmic tyrosine kinases that together form a unique family and are each essential for BCR and T-cell receptor signaling, respectively.41 Mice with a genetic deletion of SYK have a severe impairment of B-cell development at the pro-B-cell to pre-B-cell transition and lack mature B cells. The fact that ZAP70 is expressed during B-cell development and can partially substitute for a loss of SYK indicates some functional redundancy. However, ZAP70 does not appear to have unique functions in normal B cells. In CLL, ZAP70 expression is more common in U-CLL than M-CLL, correlates with a more progressive disease course, and enhances the cellular response to BCR activation and chemokines.34,35 Interestingly, the increased BCR response in ZAP70 expressing CLL cells is independent of its kinase activity and appears to be mediated by inhibition of events that terminate the signaling response, culminating in a more prolonged activation of SYK.5,34
Upon antigen binding to the BCR, LYN phosphorylates SYK, which in turn amplifies the initial BCR signal and activates the downstream signaling cascade. In addition, SYK is involved in chemokine, integrin, and Fc-receptor signaling.41 SYK is constitutively activated (pY352) in peripheral blood CLL cells in a large proportion of cases. However, there was no correlation between the degree of SYK activation and clinical or biologic features of more aggressive disease.30 One possible explanation for this could be that blood cells only partially reflect the degree of activation of signaling pathways in the tissue microenvironment.5
SYK inhibitors
Fostamatinib (R788, an oral pro-drug of the active metabolite R406) is an ATP-competitive kinase inhibitor that also inhibits a number of other kinases.42 Whereas SYK inhibitors were initially developed for use in inflammatory diseases,42 preclinical in vitro and in vivo studies identified SYK as a promising target for the treatment of CLL and other B-cell malignancies.30,31,43–45 Treatment of CLL cells with fostamatinib in vitro inhibits BCR and integrin signaling, antagonizes the protective effect of stromal cells, reduces migration to chemokines and adhesion to stromal components, and induces a moderate degree of apoptosis.19,30,31 Thus, inhibition of SYK antagonizes BCR-dependent and BCR-independent prosurvival effects in the tissue microenvironment. The therapeutic potential of SYK inhibitors in B-cell malignancies is supported by studies in mouse models; fostamatinib prevents disease progression both in TCL1 transgenic mice (in which antigen-dependent selection appears to play a similar role as in human CLL) and in a non-Hodgkin lymphoma model that depends on cooperation between Myc and BCR-derived signals.44,45
Clinical experience
The first clinical trial of a SYK inhibitor used fostamatinib in a phase 1/2 study in patients with relapsed/refractory non-Hodgkin lymphoma and CLL.46 The phase 1 part established a dose of 200 mg oral bid for phase 2 testing. The dose-limiting toxicity was a combination of diarrhea, neutropenia, and thrombocytopenia. In the phase 2 portion of the trial, the most common adverse events were reversible cytopenias, fatigue, diarrhea, and hypertension. Of 11 patients with CLL, 6 (55%) achieved a partial response (PR). The response rate in CLL was the highest, ahead of DLBCL (22%), mantle cell lymphoma (11%), and follicular lymphoma (10%). So far, no follow-up studies of fostamatinib in B-cell malignancies have been initiated, although a recently completed randomized phase 3 study in rheumatoid arthritis showed significant activity and good tolerability of the drug.47 Newer more potent and more specific SYK inhibitors have been developed that also demonstrate promising preclinical activity in CLL.48
Targeting PI3K
The PI3K pathway is a key “hub” linking many signaling pathways to cellular growth, proliferation, and survival.49,50 PI3K consists of 2 components, p85, and p110. The regulatory subunit p85 binds via SH2 domains to phosphotyrosine motifs on receptor tyrosine kinases or adaptor molecules. When p85 binds to phosphotyrosines, the catalytic p110 subunit is activated and phosphorylates phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2 (PIP2)) to generate PI(3,4,5)P3 (PIP3). PIP3 serves as a docking site for cytoplasmic kinases that include BTK and AKT, leading to the assembly of a functional signaling complex (Figure 1). The PI3K isoforms α and β are ubiquitously expressed, whereas the PI3Kδ isoform is primarily expressed in leukocytes. PI3Kδ is essential for antigen-induced BCR signaling, and its deletion in mice leads to virtual absence of B1 B cells and marginal zone B cells but not of follicular B cells. In contrast, PI3Kα and PI3Kδ fulfill redundant functions in tonic BCR signaling and constitutively active PI3Kα is sufficient to rescue B cells that have lost BCR expression.49,51,52 Upon antigen-dependent BCR activation, PI3Kδ generates PIP3 that recruits BTK, PLCγ2, and AKT, which in turn amplify the signal and mediate the functional effects of BCR activation. Although BTK can be tyrosine phosphorylated in the absence of PI3Kδ, downstream effects such as calcium mobilization and cell proliferation are severely reduced. PI3Kδ also plays a role in the transduction of costimulatory signals originating from BAFF, CD40, and TLR pathways. In addition to its role in B cells, PI3Kδ promotes maturation and expansion of CD4 T-cell subsets and of T-regulatory cells.50
PI3K inhibitors
GS-1101 (CAL-101) is a highly selective inhibitor of the PI3Kδ isoform that induces apoptosis in CLL cells in vitro and inhibits the supportive effect of many microenvironmental factors, including coculture with “nurse-like cells,” activation of the BCR, and of CD40L, BAFF, TNF-α, or fibronectin.53–55 GS-1101 inhibits activation of AKT and ERK, down-regulates MCL1, and inhibits secretion of cytokines and chemokines in vitro and in vivo.53–55 In particular, the chemokines CCL3 and CCL4, which are up-regulated in CLL cells stimulated through the BCR,5,56 rapidly decreased in the serum of CLL patients treated with GS-1101.55 Although GS-1101 does not decrease T-cell viability directly, it can inhibit the secretion of inflammatory and antiapoptotic cytokines.54
Several PI3K inhibitors are in preclinical and early clinical studies in hematologic malignancies. The PI3Kα inhibitors PIK-90 and PI-103 were more effective than PI3Kδ or PI3Kβ/δ specific inhibitors at inhibiting CLL cell migration to CXCL12 and in antagonizing stromal cell-mediated survival signals.57 Similarly, rigosertib, a PI3Kα/β inhibitor in advanced clinical testing for myelodysplastic syndromes, induced apoptosis in CLL cells cultured in contact with stromal cells.58 Isoform selective targeting of the PI3K pathway appears to be an attractive option to prevent unwanted effects on normal cells. However, in transformed cells, the dominant role of a specific isoform may be lost and different isoforms can assume redundant functions.59 Whether this will give rise to treatment failure remains to be defined.
Clinical experience
A phase 1 study of GS-1101 in hematologic malignancies established a dose of 150 mg twice a day for future studies. Fifty-four patients with CLL were enrolled. OR by International Workshop on Chronic Lymphocytic Leukemia (IWCLL) criteria60 was 26%.61,62 However, 80% of patients had a reduction in lymphadenopathy by ≥ 50%; many of these patients did not meet criteria for response by IWCLL criteria because of a transient increase in the absolute lymphocyte count (ALC). PFS was not reached at > 11 months, and responses were independent of classic risk factors and also seen in patients with 17p deletion. Grade ≥ 3 adverse events included pneumonia (24%), neutropenia (24%), thrombocytopenia (7%), neutropenic fever (7%), anemia (6%), and ALT/AST increase (6%). The relatively high rates of infection may reflect the heavily pretreated patient population. Other side effects were generally mild. GS-1101 reduced AKT activation in CLL cells of patients on treatment and normalized plasma concentrations of CCL3 and CCL4.55 GS-1101 is currently studied in various combinations; the combination with rituximab and/or bendamustine for previously treated CLL achieved an OR of 71% by IWCLL criteria.
Targeting BTK
BTK is a cytoplasmic tyrosine kinase of the Tec family that is essential for BCR signaling.63 Loss-of-function mutations in BTK block B-cell maturation at the pre-B-cell stage and cause X-linked agammaglobulinemia (Bruton agammaglobulinemia), which is characterized by the virtual absence of B cells and immunoglobulins and results in recurrent bacterial infections. Genetic deletion of BTK in mice causes the less severe xid phenotype characterized by reduction in peritoneal B1 cells and marginal zone B cells, 2 B-cell populations dependent on continued antigen-induced BCR signaling. BTK is expressed in B cells from early precursors to mature B cells and in myeloid cells but not in plasma cells or T lymphocytes. However, BTK does not appear to have essential functions outside of the B-cell compartment. BTK is recruited into the signaling complex at the plasma membrane via docking of its pleckstrin homology domain to PIP3. Mutations in the pleckstrin homology domain of BTK cause a severe B-cell defect despite normal expression of the protein, indicating the crucial importance of its proper localization. BTK is required for BCR induced calcium release, cell proliferation, and activation of the NF-κB pathway. In addition to its role in BCR signaling, BTK functions in several signaling pathways that may also be activated in the tissue microenvironment (Table 1).63
First indications that targeting BTK may be a promising strategy to treat some B-cell malignancies came from 2 complementary studies: the first, a genetic interference screen in ABC-DLBCL cell lines, identified BTK as essential for tumor cell growth and survival29 ; and the second demonstrated that B-cell lymphomas that spontaneously arise in dogs responded to the investigational BTK inhibitor ibrutinib (PCI-32765).64
BTK inhibitors
Ibrutinib is an orally available irreversible and specific inhibitor of BTK.63,65 It binds covalently to Cys-481 and thereby inhibits BTK for > 24 hours despite its short half life. Several investigators have shown that ibrutinib can inhibit survival, proliferation, and migration of CLL cells in in vitro models of the tumor microenvironment.66–68 Herman et al reported that ibrutinib not only inhibited BCR signaling but also disrupted the protective effect of stromal cells, inhibited CD40, BAFF, TLR, and cytokine signaling.67 In addition, ibrutinib blocked secretion of cytokines from activated T cells while not affecting their survival. Studies by Ponader et al showed that ibrutinib inhibited survival and proliferation of CLL cells in vitro in response to BCR activation or coculture with “nurse-like cells.”68 Ibrutinib inhibited secretion of CCL3 and CCL4 by CLL cells, and patients treated with ibrutinib showed a rapid decrease in serum concentrations of these chemokines, suggesting that CCL3 and CCL4 serum levels could become valuable biomarkers of drug activity.68 Furthermore, ibrutinib inhibited migration to CXCL12 and CXCL13 and decreases adhesion to stromal elements, such as fibronectin and VCAM1.66,68 Ibrutinib was also effective in inhibiting disease progression in TCL1 mice and inhibited activation and proliferation of CLL cells xenografted into immune-suppressed mice.68,69 AVL-292 is another orally available irreversible inhibitor of BTK that recently entered clinical testing.70 Furthermore, as previously mentioned, dasatinib not only inhibits LYN but also BTK.38
Clinical experience
The initial phase 1 dose escalation study of ibrutinib reported responses in 60% of patients with various B-cell malignancies. In the 14 patients with CLL, the OR was 79%, including 2 complete responses. Using a bioprobe assay to measure effective BTK inhibition, a dose of 420 mg was found to result in > 90% inhibition of BTK and was chosen for further study.71 A phase 1b/2 study of ibrutinib in CLL enrolled 2 cohorts: treatment-naive patients > 65 years and relapsed/refractory patients. In the latter cohort, best responses were PR in 66% and complete responses in 1 of 61 patients studied, with no difference between dose levels.72 An additional 23% of patients had a reduction in lymphadenopathy > 50% but not sufficient reduction in ALC to fulfill criteria of PR. OR > 60% were seen in all risk groups, including in patients with 17p deletion or who were fludarabine refractory. The 12-month PFS was 86%. In the cohort of untreated elderly patients, 73% of patients achieved a PR by IWCLL criteria and the PFS at 12 months was estimated to be 93%.73 Ibrutinib was well tolerated, and the most common side effects were diarrhea, nausea, fatigue, echymosis, upper respiratory tract infections, muscle spasms, arthralgia, peripheral edema, and pyrexia. Grade 3 or 4 cytopenias were seen in < 10% of patients. Grade ≥ 3 infections were seen in 26% of patients in the relapsed cohort, which is comparable to the rate of pneumonias on GS-1101 and may reflect the infection risk in this high-risk patient population. Interestingly, preliminary data suggest that ibrutinib neither depletes normal B cells nor significantly reduces immunoglobulin levels.63,71
Considerations and outlook on the role of kinase inhibitors in CLL
Kinase inhibitors work: must we and can we know why they work in CLL?
Whether the therapeutic effects of the kinase inhibitors discussed here can be attributed to the inhibition of BCR signaling is not easy to ascertain (Table 2). First, in the absence of direct genetic proof, the importance of the BCR in CLL is based on circumstantial evidence. However, functional studies with CLL cells in vitro, ongoing activation of CLL cells through the BCR in vivo, clinical observations, and genetic evidence in lymphoma and mouse models strongly implicate the BCR as the pivotal pathway in pathogenesis and disease progression. A second confounding issue is that, in addition to the BCR, many pathways with important roles in B-cell biology activate SYK, BTK, and the PI3K pathway (Table 1). Whether these pathways function primarily as costimulatory pathways to BCR activation or are sufficient for CLL cell expansion in vivo is not clear. Nevertheless, it appears reasonable to classify inhibitors of SYK, BTK, and PI3Kδ as “BCR inhibitors,” acknowledging that it may well be that these kinase inhibitors are efficacious exactly because they simultaneously target several pathways that promote B-cell survival.67,68 Understanding more about the mechanistic aspects of BCR inhibitors will be especially important in regards to resistance mechanisms and the design of combination therapies.
“Redistribution lymphocytosis” and the need for revised response criteria
Clinical responses to SYK, BTK, and PI3K inhibitors in CLL are characterized by a significant decrease in lymphadenopathy and splenomegaly within weeks that is frequently accompanied by a rise in the ALC. The rise in circulating tumor cells can be rapid, with a lymphocyte doubling time measured in weeks and can increase the ALC to > 5-fold baseline. This transient lymphocytosis typically takes several months to resolve. In the face of improvement in other clinical parameters and a decrease of tumor burden in lymphoid organs, the rising ALC does not indicate disease progression, and there is currently no evidence that it is associated with morbidity. Thus, patients can and should be maintained on the drug. However, because of the lymphocytosis, patients are often slow to reach response criteria by IWCLL guidelines despite significant reductions in both lymphadenopathy and splenomegaly and overall symptomatic improvement. Paradoxically, some patients who obviously benefit from the treatment may even fulfill classic criteria of progressive disease.60 Thus, response criteria need to be adapted to capture the clinical benefit of these novel agents and account for the different biology of CLL treated with targeted agents.74
The treatment-induced lymphocytosis is thought to be caused by a change in cell trafficking and, at least in part, may be a consequence of attenuated BCR signaling. Because BCR activation increases CLL cell migration to chemokines and enhances integrin-mediated adhesion of CLL cells to VCAM1 and fibronectin, its inhibition can reduce homing to and retention of CLL cells in the tissue microenvironment.31,66 In addition, independent of the effect on BCR signaling, BCR inhibitors may directly attenuate response to chemokines and integrins, thereby further disrupting cell trafficking and adhesion.19,48,54,55,67,68 Thus, BCR inhibitors not only disrupt these critical signaling pathways directly, but in addition may dislodge CLL cells from a protective microenvironment and sensitize them to therapeutic intervention. Although the initial lymphocytosis is common, some patients have rapid resolution of not only lymphadenopathy and splenomegaly but also of leukemic disease. The reason for these differences in response patterns remains to be determined. Interestingly, despite significant overall reductions in tumor burden, reports or indications of tumor lysis syndrome with BCR inhibitors are notably absent.
Redefining treatment goals and the development of rational combination therapies
As final results of single-agent therapy are only just emerging, combination therapies are being explored. In addition to combinations of BCR inhibitors with chemotherapeutic agents and monoclonal antibodies, preclinical data suggest intriguing novel combinations. For example, the BCL2 inhibitor navitoclax is a well-tolerated oral agent that induced PRs in 35% of relapsed CLL patients with a median PFS of 25 months.75 High MCL1 expression and a low BIM to MCL1 ratio were predictive of inferior response. In particular, disease in the bone marrow was less responsive to navitoclax, which may be the result of up-regulation of MCL1 in CLL cells in contact with stroma and a decrease in BIM.32,76 Inhibition of signals from the microenvironment can reduce MCL1 expression and up-regulate proapoptotic BIM, providing a rationale for combinations of kinase inhibitors with BH3 mimetics, such as navitoclax.30,76 Another attractive combination could be an immune-stimulatory agent, such as lenalidomide with a BCR inhibitor. At least in ABC-DLBCL models, there is direct evidence for synergy between ibrutinib and lenalidomide.77 In contrast, lenalidomide-induced immune stimulation in CLL has been shown to depend on PI3Kδ and was inhibited by GS-1101.78 Although inhibition of inflammatory cytokine secretion might improve its tolerability, the combination could also decrease the clinical benefit of lenalidomide, given evidence that its efficacy in CLL depends on the activation of an adaptive immune response.79,80 The clinical success of such combinations may therefore depend on a more detailed understanding of differences between the various BCR inhibitors in regards to what pathways are inhibited and the resulting impact on cellular networks.
The development of relatively well-tolerated and highly active kinase inhibitors raises the question about their future role in the treatment of CLL. Will they best be used in combination with chemotherapy to maximize cytotoxicity in curative intent or could they provide means for the chronic suppression of the leukemic clone? The ability of the BCR inhibitors to disrupt the protective effect of the tumor microenvironment may argue for their use as “sensitizing agents” in combination therapies. On the other hand, the impressive single-agent activity suggests a possibility that some of these agents may be valuable as stand-alone therapy or that they could become the backbone of chemotherapy-free combination regimens. In due course, BCR inhibitors may also become an option for early intervention in high-risk patients. Treatment goals and preferred strategies will become clearer as we learn more about duration of response, mechanisms of resistance, tolerability of chronic use, and the differences between the various kinase inhibitors.
This article was selected by the Blood and Hematology 2012 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2012. This article is reprinted with permission from Blood. 2012; Volume 120.
Acknowledgments
The author thanks Andrew Lipsky for critical reading of the manuscript and his colleagues for helpful discussions.
A.W. is supported by the intramural research program of the Heart, Lung, and Blood Institute, National Institutes of Health.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Off-label use of kinase inhibitors including ibrutinib (PCI-32765), GS-1101 (CAL-101), fostamatinib in the treatment of CLL. Navitoclax and lenalidomide in the treatment of CLL and in combination with the kinase inhibitors.
Correspondence
Adrian Wiestner, Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Dr, Bldg 10, CRC 3-5140, Bethesda, MD 20892-1202; Phone: 301-594-6855; Fax: 301-594-1290; e-mail: wiestnera@mail.nih.gov.