Abstract
In recent years, research in genomics has resulted in the rapid uncovering of the molecular pathogenesis of acute myeloid leukemia (AML). The identification of the genetic determinants of response to standard—but also to experimental—treatment is increasingly used for patient counseling, to guide clinical decision making, and for resource-efficient care provision at diagnosis, during consolidation treatment and follow-up, and after relapse. Gene mutations now allow us to explore the enormous diversity among cytogenetically defined subsets of AML, in particular the large subset of cytogenetically normal AML. Nonetheless, there are several challenges in evaluating the prognostic value of a specific mutation in the concert of the various concurrent mutations and determining the relative prognostic value of the genetic profile during the disease course. In particular, changes in the genetic profile in relapse compared with that at diagnosis will increasingly affect the treatment strategy at relapse, but also will give us the possibility of learning which treatment strategy during frontline therapy is best to prevent them.
Introduction
Acute myeloid leukemia (AML) is a genetically very heterogeneous disorder with an incidence of 3 to 4 per 100 000 men and women per year. It is characterized by the accumulation of somatically acquired genetic changes in hematopoietic progenitor cells that alter normal mechanisms of self-renewal, proliferation, and differentiation. Outcome is influenced by various factors, including patient features such as age, comorbidities, and performance status and disease characteristics of which the genetic profile of the disease is the most important. According to the recommendations from an international expert panel, on behalf of the European LeukemiaNet (ELN), AML can be grouped into 4 risk groups as shown in Table 1.1
Based on registry data, the median age at diagnosis of patients with AML ranges from 66 to 71 years (Surveillance Epidemiology and End Results [SEER] Cancer Statistics Review 1975-2009)2 and the proportion of patients receiving intensive chemotherapy decreases with increasing age.3
In patients considered suitable for intensive induction therapy, the combination of an anthracycline and cytarabine (“7 + 3”) remains the standard of care. Complete remission (CR) can be achieved in 65% to 75% of younger adult patients (≤ 60 years) and in approximately 40% to 60% of older patients (> 60 years). The poor CR rate and overall survival (OS) in older AML patients is attributed to a variety of factors, including inherently poor biology (especially a higher incidence of poor-risk karyotypes), comorbidities, and an age-related functional decline.
In patients ineligible for intensive chemotherapy, the spectrum of treatment options is limited and includes best supportive care (with hydroxyurea), low-dose cytarabine, and the hypomethylating agents decitabine or azacitidine (20%-30% BM blasts). Using such low-dose therapy, CR can be achieved in 10% to 30% of patients and the OS at 3 years is approximately 5%.4
In patients who achieve a CR after induction chemotherapy, some postremission therapy (PRT) is required to prevent relapse. Although the value of PRT in the older patients continues to be debated, in younger patients, the choice for consolidation is based on genetic and molecular features and can range from high-dose cytarabine to allogeneic hematopoietic stem cell transplantation (allo-HSCT), with a 5-year OS rate of 40% to 45%; OS in older patients still remains poor at < 10% after 5 years.1
Considering the genetic heterogeneity of AML, it is very unlikely that already established or new agents including combination therapies are equally effective in all genetic subgroups. Therefore, the identification of the genetic determinants of response to treatment, including achievement of a CR and survival end points, is of high importance for individual patient counseling and resource-efficient care provision. This has been demonstrated clearly in patients with acute promyelocytic leukemia.5 The concentration on one distinct genetically defined subgroup of AML in multinational, multicenter clinical trials fostered the identification of a clinically highly relevant new standard in low- and intermediate-risk patients.6
The goal of this review is to highlight the importance of genomics in clinical decision making at different time points during an individual AML patient's disease course, including: (1) at diagnosis with regard to classification of the disease and prognostication on achievement of a CR after induction therapy, (2) during PRT and follow-up with respect to the choice of the most appropriated strategy in first CR based on pretreatment markers (ie, intensive chemotherapy or allo-HSCT), and (3) prognostication on CR achievement after salvage therapy after relapse. In addition, genomics are increasingly entering the inclusion/exclusion criteria of clinical trials; in particular, those with genotype-adapted and/or targeted treatment approaches (eg, www.clinicaltrials.gov identifier NCT00850382, NCT01238211, NCT01830361, NCT00893399, and NCT01237808). In this review, only markers with strong prognostic impact are discussed; that is, markers that have been demonstrated in studies to play a prognostic role in CR achievement and survival end points and therefore may have the potential to influence the decision-making process.
Diagnostic work-up/disease classification
Based on the revised World Health Organization (WHO) publication WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues,7 a total of 7 entities are defined within the subgroup “AML with recurrent genetic abnormalities.” In this category, AML characterized by specific fusion genes are grouped (Table 2) and “AML with t(8;21)(q22;q22); RUNX1-RUNX1T1” and “AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11” are considered as AML regardless of BM blast counts. All other entities in this category require the presence of at least 20% BM blasts at diagnosis based on morphology. Two provisional entities defined by the presence of gene mutations were added to this category: AML with mutated NPM1 and AML with mutated CEBPA. The category “AML with mutated NPM1” is by far the largest subgroup defined by genomics, with a high incidence in both young and older AML patients.8-10 However, the association with cooperating genetic mutations, in particular FLT3-internal tandem duplication (ITD), seems to be age dependent, with a significantly higher incidence of the genotype NPM1mut/FLT3-ITDneg in older compared with younger patients.8
New insights have been provided for AML with mutated CEBPA. Several studies have shown convincingly that AML with double mutant CEBPA (CEBPAdm) can be distinguished from AML with single mutant CEBPA with respect to biological and prognostic features.11-14 The favorable prognostic impact of mutant CEBPA that was demonstrated previously in several studies can be attributed to the subtype of AML with CEBPAdm.11-14 Therefore, several investigators have suggested restricting the provisional entity “AML with CEBPA mutations” to those with biallelic mutations.
RUNX1 mutations have been reported to occur with an incidence of 5.6%15 to 13.2%,16 predominantly in patients with intermediate-risk cytogenetics. In patients with cytogenetically normal (CN) AML, the incidence seems to increase with higher age, with an incidence of 8% in younger patients compared with 16% in older patients.17 Interestingly, RUNX1 mutations are almost mutually exclusive of other disease-defining genetic aberrations such as NPM1, CEBPAdm, CBFB-MYH11, RUNX1-RUNX1T1, and PML-RARA.15-17 In addition, RUNX1 mutations are characterized by a distinct gene expression pattern,15,16 and monoallelic germline mutations have been reported in rare cases of familial platelet disorder with predisposition to AML,18 further supporting the idea of a separate disease entity.
A recent landmark publication by the Cancer Genome Atlas Research Network on the genomic and epigenomic landscapes of adult de novo AML reported results from next-generation sequencing performed in 200 AML patients (n = 50 whole-genome sequencing, n = 150 whole-exome sequencing).19 The investigators identified 23 significantly mutated genes and another 237 gene mutations were found in 2 or more samples. The median number of mutated genes in coding sequences was 13 (range, 0-51). The investigators proposed a classification of gene mutations into 9 categories based on their biological function, with 199 of the 200 analyzed patients having at least one mutation in 1 of these categories (Table 3). These findings will probably influence the future disease classification system.
Prognostication of response to induction therapy
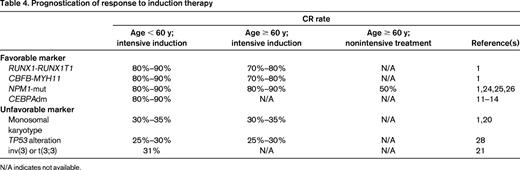
The achievement of CR after induction therapy is a commonly accepted prerequisite for long-term survival and cure. CR rates vary widely in the different prognostic groups (Table 1), from 80% to 95% in the favorable risk group to only 32.5% in patients with monosomal karyotype20 and 31% in patients with inv(3)21 ; both aberrations are categorized in the adverse-risk group (Table 4). If no CR is achieved after induction therapy, the probability of dying from AML is as high as 75% during 1 year.22 In this situation, molecular and cytogenetic markers may help to guide patients and their families through the risks and benefits of induction chemotherapy.
Genetic mutations also aid in predicting response. NPM1 mutations, one of the most frequent gene mutations (occurring in 25%-35% of all adults with AML)23 have consistently been reported as a favorable prognostic factor for CR achievement, with CR rates of 90% and higher in younger patients either as a single marker or as combined genotype, NPM1-mut/FLT3-ITDneg.1,23 CR rates after intensive induction therapy in older patients with NPM1 mutations are nearly as high as in younger patients, with CR rates of 80% to 90%.9,10 Furthermore, even in older patients who received nonintensive chemotherapy (low-dose cytarabine and etoposide) in an ongoing clinical trial (www.clinicaltrials.gov identifier NCT01237808), the NPM1 mutation was associated with a CR rate of 40% to 50% (R.F.S., unpublished data, May 2013). Whether NPM1 mutations may also serve as a predictive marker remains unknown. The addition of all-trans retinoic acid to intensive induction therapy24 and the intensification of daunorubicin within a standard “7 + 3” regimen25 resulted in significantly higher CR rates, especially in AML with the NPM1 mutation. Whether FLT3-ITD adds value in prognostication of CR rate on the background of NPM1 is still a matter of debate. Schneider et al showed no impact of FLT3-ITD on CR rates in either NPM1-mutated or NPM1 wild-type AML.26 However, specific biological characteristics of FLT3-ITD, such as the FLT3-ITD mutant/wild-type ratio, may add prognostic value in that patients with high ratios have a significantly lower CR rate irrespective of the NPM1 status.27 AML with CEBPAdm accounts for 3% to 6% of adult AML and the frequency decreases with increasing age.8,9,11-14 In this subgroup of patients, CR rates of 85% to 92% have been reported; however, these results are mainly based on younger adults (Table 4).
TP53 alterations including mutations and losses are found in approximately 70% of AML with complex karyotype.28 TP53 alterations predict for very low CR rates (less than 30%) and have been shown to be an independent poor prognostic factor among the subgroup of AML with complex karyotype. Whether more novel agents, such as the demethylating agents, may improve the dismal outcome of AML with TP53 alterations is unknown.
Several other gene mutations have been identified; however, none appears to have a strong impact on the probability of achieving a CR. Such markers include mutations of the DNMT3A (incidence 17%-25%),29-32 RUNX1 (6%-13%),15-17 IDH1/2 (20%-25%),25,33,34 TET2 (8%-21%),35,36 and WT1 (10%-15%) genes (Table 3).23
Prognostication of relapse and OS
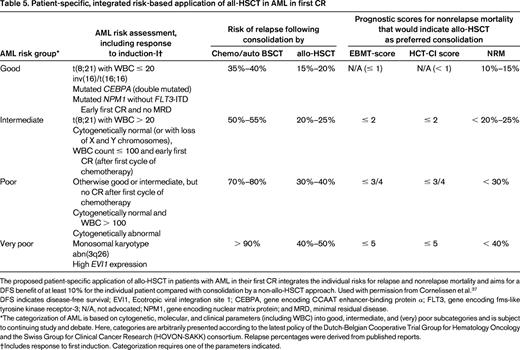
Various types of PRT, including high-dose cytarabine and allo-HSCT, have been evaluated with the aim of preventing relapse and improving OS. Although allo-HSCT is considered as the PRT with the strongest antileukemic effect, the benefit of allo-HSCT on OS may be compromised by nonrelapse treatment-related mortality. The ELN AML Working Party has proposed an integrated risk-adapted approach for patients in first CR taking into account: (1) the risk of relapse after intensive chemotherapy compared with allo-HSCT, (2) treatment-related mortality of allo-HSCT, and (3) patient and transplant-specific parameters such as comorbidity, donor type, and age reflected by the hematopoietic cell transplantation comorbidity index (HCT-CI) and European Group For Blood and Marrow Transplantation (EBMT) scores (Table 5).37 According to this recommendation, AML with RUNX1-RUNX1T1 (only with pretreatment WBC count ≤ 20/nL), CBFB-MYH11, NPM1mut/FLT3-ITDneg, and CEBPAdm were grouped into the good-risk category, whereas AML with monosomal karyotype, abn(3q), and those with high EVI1 expression were grouped into the very-poor-risk category.37 For the 2 remaining categories in between (ie, intermediate and poor risk) a combination of cytogenetics and response to initial chemotherapy are used for grouping. Based on the integrated approach, allo-HSCT represents the most appropriated PRT in patients with low HCT-CI and EBMT scores in the categories intermediate, poor, and very poor. However, the recommendations become more complex, especially in the intermediate- and poor-risk groups, with rising HCT-CI and EBMT scores. Two major shortcomings of these recommendations are that several gene mutations with high incidences, especially in the categories intermediate and poor (eg, DNMT3A-mut, TET2-mut, IDH1/2-mut, RUNX1-mut, WT1-mut), are not integrated and, even more important, the interaction of genetic aberrations are only poorly reflected. In the following, these 2 aspects are highlighted according to risk group proposed by the ELN AML Working Party.
Good risk
In patients with core binding factor (CBF) AML, KIT mutations have been associated with an increased relapse rate (Table 5).23,38 However, based on a recent report on AML with inv(16)/t(16,16), this unfavorable impact on relapse rate does not translate into an inferior survival. In contrast, AML with inv(16)/t(16,16) harboring additional FLT3 mutations including FLT3-ITD and FLT3-TKD was associated with a strong negative impact in multivariable analysis on OS.38 However, presently, due to the inconsistencies in the available data, cooperating gene mutations in CBF-AML should not be used to guide treatment decisions. In AML exhibiting the genotype NPM1-mut/FLT3-ITDneg two reports from cooperative study groups showed a negative impact of cooperating IDH1/2 mutations on relapse-free survival and OS.33,34 In contrast, Patel et al reported on a favorable impact of the genotype NPM1-mut/FLT3-ITDneg only if cooperating IDH1/2 mutations were present.25 Such opposed effects of genotypes on outcome highlights statistical shortcomings of retrospective molecular studies.
Further conflicting results have been reported on the prognostic value of TET2 mutations in AML with NPM1-mut/FLT3-ITDneg or CEBPAdm.35,36 Metzeler et al demonstrated that in ELN favorable-risk patients with CN-AML who have a CEBPAdm and or NPM1mut/FLT3-ITDneg, TET2 mutated patients did poorly on all survival end points.36 In that analysis, TET2 mutations were significantly more frequent in older compared with younger patients. Although multivariable analysis revealed an independent impact of TET2 mutations, age may be an important confounding factor. This is supported by the report from Gaidzik et al focusing on a large cohort of homogeneously treated younger adults.35 In that study, TET2 mutations had no prognostic impact in the whole group or in any of the subgroups, including those defined by the genotypes NPM1-mut/FLT3-ITDneg and CEBPAdm. Therefore, the prognostic value of TET2 mutations, at least in younger patients, is limited; in older patients, a confirmatory study of the results from Metzeler et al is needed.
Intermediate risk
The intermediate-risk category (Table 5) of the recommendations comprises mainly patients with CN-AML who achieve a CR after induction therapy.
DNMT3A has been found to be mutated frequently in AML with normal karyotype (30%-35%).29-32 Two studies have demonstrated that DNMT3A mutations are independently associated with poor OS.29,30 However, patients exhibiting a DNMT3A mutation were significantly older in both studies, so, again, age may be an important confounding factor in these analyses. Marcucci et al reported on a differential prognostic effect of DNMT3A mutations in older versus younger patients according to the affected codon; older patients with DNMT3A mutations in codon R882 in exon 23 had an inferior outcome, whereas younger patients with DNMT3A mutations other than R882 did worse.30 In the largest analysis so far published on 1770 young adults, DNMT3A mutations had no consistent impact on survival end points in the entire group.32 However, in subgroup analyses, DNMT3A mutations were found to be associated with a unfavorable prognosis in the ELN molecular-unfavorable subgroup (Table 1) of CN-AML.
Approximately two-thirds of RUNX1 mutations are found in CN-AML and have been associated with a very unfavorable prognosis in both young and elderly patients.15,17 Gaidzik et al reported a dismal outcome for all survival end points in patients with RUNX1 mutations after intensive PRT compared with allo-HSCT in first CR.15 Therefore, the question arises whether the presence of RUNX1 mutations should in the future be categorized as poor risk or even very poor risk according to the ELN classification (Table 1).15
Poor and very poor risk
Patients categorized in the poor- or very-poor-risk group (Table 5) have per se a dismal prognosis, and most of these patients should be offered an allo-HSCT if a CR is achieved.37 TP53 alterations are closely associated with a complex and in particular also with a monosomal karyotype,28 so the majority are already categorized in the very-poor-risk group. However, if a CR is achieved, allo-HSCT should be offered if possible. Futures studies will be needed to determine whether maintenance therapy, such as therapy with hypomethylating or other novel agents, may improve survival of those patients who are unable to proceed to allo-HSCT. Unfortunately, a large number of older AML patients tend to have poor-risk karyotypes and are ineligible for intensive induction therapy and allo-HSCT. Future studies in this patient population are imperative.
Prognostication in first relapse
Approximately half of younger patients and 90% of older patients relapse and these relapses often appear to be associated with clonal genetic evolution. Whole-genome sequencing studies by Ding et al have offered insights into the pathogenesis of relapse and demonstrated that the founding clone in the primary AML gains mutations and evolves into the relapse clone and a subclone of the founding clone survives initial therapy, gains additional mutations, and expands at relapse.39 In both scenarios, it may be helpful for the clinician to know the genetic background of the disease at relapse. However, most existing data on prognosis after relapse are based on the pretreatment cytogenetic and molecular genetic profile40 and the general notion is that a second CR is rarely achieved.41 Younger adults (age 16-49 years) who relapsed after intensive consolidation chemotherapy had a 55% chance of achieving a second CR with highest rates of 82% in the pretreatment favorable-risk group comprising t(8;21), t(15;17), and inv(16).40 In addition, relapsed patients exhibiting the pretreatment favorable genotype NPM1-mut/FLT3-ITDneg also had a high second CR rate of 66%, whereas relapsed patients exhibiting a FLT3-ITD at diagnosis only had a 35% second CR rate irrespective of an additional NPM1 mutation. Based on these data, the current practice to postpone allo-HSCT in young adults to second CR in patients with favorable genetics seems reasonable because results of allo-HSCT in second CR are comparable to those performed in first CR.40 However, these data are based on a minority of AML patients due to the age restriction of 16 to 49 years and therefore the results cannot be generalized. In addition, clonal evolution may influence the probability of achieving a second CR, which has been exemplarily shown by Krönke et al in AML with NPM1 mutations.42 In 53 patients with AML exhibiting NPM1 mutations at diagnosis who relapsed, 5 lost the NPM1 mutation while maintaining the initially already coexisting DNMT3A mutation. Two-thirds of the patients with persistent NPM1 mutation achieved a second CR, whereas none of the 5 patients who lost NPM1 responded to salvage therapy. These data show clearly that the second CR rate decreases by 25% to 30% compared with first CR rate even if the main genotype (ie, mutated NPM1) remains stable.
From a clinical point of view, it would be very helpful to know the rate of second CR after intensive chemotherapy or alternatively after tyrosine kinase inhibitor therapy as a single agent43 based on the molecular profile at relapse.
Conclusion
Progress in deciphering the molecular pathogenesis of AML and the identification of the genetic determinants of response to treatment have been impressive, and translation of these findings into the clinical decision making has been increasing in recent years. After the successful implementation of a fast molecular screening within 48 hours for FLT3-ITD, FLT3-TKD, and the fusion genes in CBF-AML and acute promyelocytic leukemia within the international Cancer and Leukemia Group B (CALGB) 10603 study in 2008, this strategy has been adopted and expanded in several study groups but also on the national health service level in France. The fast availability of the molecular disease profile prompted a large body of genotype-specific clinical trials that may change clinical practice soon. Nonetheless, given the enormous molecular heterogeneity of the disease, international collaborations are needed to open the possibility of studying large cohorts with the aim of minimizing selection bias and enabling evaluation of rare genetic aberrations. Of prime importance is the evaluation of the genetic profile at all clinically relevant time points, including at diagnosis, but of nearly comparable importance at relapse.
Disclosures
Conflict-of-interest disclosure: R.F.S. has received research funding from Pfizer, Celgene, Amgen, and Novartis and has received honoraria from Celgene, Teva, and Ambit. H.D. has consulted for Celgene, Bristol-Myers Squibb, Novartis, Ambit, Astellas, Epizyme, Arog Pharmaceuticals, and Clavis. Off-label drug use: Gemtuzumab ozogamicin, adjunct to intensive induction therapy; all-trans retinoic acid, adjunct to intensive induction therapy; sorafenib, midostaurin, quizartinib, and volasertib either as single agents or in combination with chemotherapy.
Correspondence
Richard F. Schlenk, MD, Department of Internal Medicine III, University Hospital Ulm, Albert-Einstein-Allee 23, 89081 Ulm, Germany; Phone: 49-73150045901; Fax: 49-73150045905; e-mail: richard.schlenk@uniklinik-ulm.de.