Abstract
Substantial progress has been made in the treatment of precursor B-cell acute lymphoblastic leukemia (B-ALL), but recurrent disease remains a leading cause of death in children due to cancer and outcomes for adults with B-ALL remain poor. Recently, complete clinical responses have been observed in small numbers of patients with B-ALL treated with adoptive immunotherapy using T cells genetically engineered to express chimeric antigen receptors (CARs) targeting CD19, a cell surface molecule present in essentially all cases of B-ALL. Preclinical data suggest that CARs targeting CD22, another antigen present in the majority of B-ALL cases, are similarly potent. Several clinical studies already under way will soon more clearly define the rate of response to this novel therapy in B-ALL. Further work is needed to identify optimal platforms for CAR-based adoptive immunotherapy for leukemia, to establish guidelines for managing toxicity, and to determine whether the remissions induced by this approach can be rendered durable.
Introduction
Approximately 5000 to 6000 new cases of precursor B-cell acute lymphoblastic leukemia (B-ALL) are diagnosed in the United States each year, with more than half of these cases occurring in individuals under the age of 20 years. Modern multiagent regimens have resulted in impressive cure rates of almost 90% in children with B-ALL,1 yet due to its relatively high incidence compared with other childhood cancers, B-ALL remains a leading cause of death in children due to cancer. Risk stratification based on clinical features, cytogenetics, and response to therapy have allowed tailoring of therapy in children to reduce long-term toxicity with preservation of excellent survival. However, outcomes with chemotherapy alone remain poor for some high-risk groups. Furthermore, even modern risk-adapted strategies require prolonged treatment regimens with substantial long-term morbidity.
Improvements in survival have also been achieved in other age groups, but with declining success with increasing age (5-year overall survival rates of ∼ 60% for ages 15-20 and 45% for ages 20-30 years). For adults older than 30 years at the time of diagnosis, overall survival is estimated to be 20% to 40%.2,3 Recent data indicate that the use of more intensive pediatric regimens in adults can improve outcomes somewhat,4 but the high-risk features associated with B-ALL in adults makes it unlikely that the successes seen in children will be achieved with currently available regimens. Treatment of relapsed B-ALL also remains a substantial challenge, with outcomes varying depending on several factors, including age and length of first remission. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) leads to cure in ∼ 50% of patients who achieve second complete remission.5 However, substantial numbers of patients do not successfully reach a second complete remission and therefore are not candidates for this therapy. As a result, data in which all relapses are included in the analysis irrespective of whether allo-HSCT is performed show a much more dismal overall survival for patients with relapsed ALL, despite intensive, highly toxic therapy.6 Furthermore, all salvage therapies currently available for B-ALL are associated with substantial short-term and long-term toxicity. Therefore, despite increasing success in the treatment of B-ALL over recent decades, outcomes remain poor for several subgroups and we have likely reached a point of diminishing returns with increasing intensity of standard cytotoxic regimens. Novel treatment modalities are needed.
The modern era of genomics has identified several potential molecular targets in B-ALL and the addition of bcr-abl inhibitors to standard cytotoxic regimens in Philadelphia chromosome–positive ALL markedly improve the outcome in this high-risk subgroup.7 However, more limited success has been achieved using inhibitors of other candidate targets such as JAK2,8 likely due to the multitude of pathways that contribute to oncogenesis in this disease.9 The emerging picture of B-ALL oncogenesis suggests that effective targeting of B-ALL using small-molecule tyrosine kinase inhibitors will be challenging. Another approach to improving outcomes is to target cell surface molecules using mAb-derived therapeutics. Unconjugated mAbs targeting CD20 have had impressive effects when combined with cytotoxic regimens in CD20+ B-cell malignancies, although expression of CD20 in pediatric B-ALL is limited to the Burkitt subtype, which represents a minority of cases presenting in childhood. Interestingly, recent studies have suggested higher rates of CD20 expression in relapsed B-ALL with improved outcomes reported in adults < 60 years of age with de novo CD20+ ALL (defined as expression on > 20% of the blast population) when anti-CD20 was added to an intensified chemotherapy regimen.3
Unconjugated anti-CD19 mAbs have not yet induced measureable antitumor effects10 ; however, bispecific anti-CD19 mAbs that simultaneously bind a cell surface antigen on B-ALL and CD3 on nearby T cells have shown impressive single-agent activity in ALL.11 mAbs can also be conjugated to agents that kill the tumor target directly without activating endogenous immune effectors. This approach has seen increasing success, including recent studies using an anti-CD22 mAb-derived binding domain plus a Pseudomonas-derived exotoxin that demonstrate clinical responses in a substantial fraction of patients with refractory B-ALL.12,13 Recently, a new approach that uses genetic engineering to endow T cells with receptors that bind leukemia cell surface antigens has shown increasing promise. This review focuses on this rapidly emerging field of adoptive therapy for B-ALL using immune cells genetically modified to express chimeric antigen receptors (CARs).
Optimizing CAR-based therapeutics for B-ALL
CARs are hybrid receptor constructs that contain a target recognition domain linked to an intracellular component that activates a signaling cascade in the immune effector cell. In most cases, the antigen-binding domain consists of single protein chains derived from mAb fragment variable regions (scFvs) connected by a short linker sequence, although antigen-binding domains derived from natural ligands are also under study.14,15 The signaling component has evolved greatly since the original description of a chimeric receptor by Eshhar et al in 1989,16 and much of the progress in this field in the last 20 years has resulted from an increasingly effective optimization of signaling moieties that can drive productive T-cell activation, expansion, effector function and survival. The native TCR-CD3 complex is composed of 6 separate chains (α, β, γ, δ, ϵ, and ζ), but signals generated via the ζ chain alone are sufficient to induce downstream events that are indistinguishable from those generated by an intact TCR.17 Not surprisingly, then, CARs that incorporate TCR-ζ alone as the sole surrogate TCR component induce full T-cell activation upon encounter with antigen. Therefore, TCR-ζ is now standardly used to provide signal one and thus activate T cells in the context of CAR therapy.18
Costimulatory endodomains
A fundamental tenet in immunology holds that T cells activated via the TCR alone are prone to anergy and do not mediate robust antitumor effects due to limited cytokine production, proliferation, and persistence. It is not surprising, then, that CARs incorporating TCR-ζ alone fail to mediate robust antitumor effects. Modern CARs also incorporate a costimulatory endodomain selected from a growing list of costimulatory molecules. Most commonly, the sequence incorporated into CAR constructs is derived from the CD28 endodomain, which enhances proliferative and cytokine producing capacity in vitro19 and enhances persistence in vivo in animal studies.20,21 The importance of a costimulatory endodomain in the context of CAR therapy was further demonstrated in a clinical trial of CD19-CAR–modified T cells administered to patients with non-Hodgkin B-cell lymphomas. Savoldo et al coinfused equal numbers of T cells bearing CD19-CARs with TCR-ζ alone or CD19-CARs with TCR-ζ plus a CD28 endodomain. T cells derived from each population were distinguishable based upon a noncoding element in the construct, allowing quantification of the 2 populations in vivo. The results clearly demonstrated that expansion of the T cells expressing CARs containing CD28 plus TCR-ζ expanded greater and persisted longer than CARs T cells expressing CARS with TCR-ζ alone.22 Therefore, the majority of CAR constructs currently being used in clinical trials contain both TCR-ζ and at least one costimulatory endodomain.
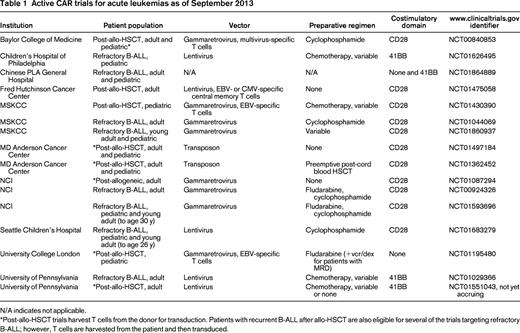
Numerous costimulatory endodomains have been incorporated into CARs, including CD28, 4-1BB,23,24 OX40,25 ICOS,26 and DAP10.15 An unanswered question in this field is whether any of these costimulatory signals are superior to the others and whether multiple costimulatory signals lead to enhanced functionality. Few direct comparisons have been made. In a study by Brentjens et al, CD28-expressing CARs were more potent in vitro compared with OX40-, 4-1BB-, and DAP10-containing CARs with respect to expansion and cytokine secretion.21 In contrast, at least one animal model of ALL demonstrated that CARs expressing 41BB-costimulatory domains had improved persistence and efficacy compared with those expressing CD28 endodomains.23 Furthermore, in some studies, the inclusion of multiple costimulatory domains led to enhanced functionality compared with CARs with single costimulatory endodomains,24 whereas in other studies, the functionality of CARs with multiple costimulatory endodomains was reduced.27 In summary, there is a consensus that incorporation of at least one costimulatory endodomain is essential for CARs to mediate meaningful antitumor effects, but it remains unclear whether any costimulatory endodomain is superior to the others. Most of the current clinical trials targeting B-ALL (Table 1) with anti-CD19 CAR-modified T cells are single-arm studies that administer cells expressing either CD28 endodomains or 4-1BB endodomains, thus precluding direct comparison. A clinical trial administering mixed populations of anti-CD19-CAR T cells expressing either CD28 or 4-1BB endodomains to individuals with refractory chronic lymphocytic leukemia or indolent lymphoma is currently under way (www.clinicaltrials.gov identifier NCT00466531) and may provide answers to this important question. In this trial, however, the vectors used for T-cell transduction and the scFv constructs also vary, potentially limiting the interpretation of the results.
Antigen selection
The fundamental basis for the promise of CAR-based therapies for B-cell precursor B-ALL comes from the fact that several molecules highly expressed on the surface of B-ALL blasts are consistently expressed across a variety of subtypes, are expressed at high levels on all malignant cells, and have nonmalignant tissue expression restricted to B-lineage cells. CD19 is the target antigen for which all of the published clinical data for CAR therapy in ALL exist. However, as noted in the Introduction, CD22 is also highly expressed on the majority of ALL cases, and anti-CD22–based immunotoxins induced antileukemic effects in early clinical trials with acceptable toxicity. Furthermore, in some cases, immune escape with CD19− blasts has been observed after CD19-targeted immunotherapy, both after CD19-targeted bispecific antibody therapy11 and after CD19-CAR–based therapy.28 Therefore, there has been substantial interest in generating a CAR capable of targeting CD22 in B-ALL. The emerging immunotoxin experience demonstrated that incorporation of binding proteins with higher affinity enhanced efficacy, because a first-generation anti-CD22 immunotoxin incorporating an scFV that was less avid showed lower rates of clinical activity compared with a second-generation anti-CD22 immunotoxin incorporating an affinity-enhanced scFv targeting the same distal epitope.12,13 However, when comparing the efficacy of CARs generated from these 2 scFvs, enhanced affinity conferred no advantage.27 Furthermore, neither scFv targeting the distal epitope showed powerful antileukemic effects, whereas a third scFv of similar affinity but targeting a more proximal epitope was clearly superior.27 These results demonstrate that CD22 is an alternative target for CAR-based therapy of B-ALL and demonstrate that epitope selection may be a critical factor affecting the potency of CARs (Figure 1). They also align with results from other studies that have concluded that, beyond a certain threshold, affinity is not a major factor in determining the efficacy of CAR therapy29 and provide further evidence for the notion that membrane proximal epitopes are preferred targets for CAR therapy.30,31 The importance of epitope location in modulating CAR activity also raises the larger issue of identifying appropriate “spacing” for individual CARs that allow for efficient binding between the CAR and the tumor cell surface target. In some cases, hinges incorporated into the CAR structure to modulate CAR size can substantially improve CAR activity and studies are under way to develop predictive algorithms for predicting the optimal size of such “spacer domains.”

Bioluminescent imaging results. NSG-immunodeficient mice were inoculated on day 0 with 5 × 105 NALM-6-GL cells and then, on day 3, received 1.5 × 107 CAR-modified T cells as designated. Mock recipients received 1.5 × 107 total cells. Transduction efficiency was ∼ 40% in both CAR groups. Mock-transduced cells were activated and expanded but were not exposed to viral vector. Bioluminescent imaging using standard techniques at the designated time points demonstrates clearance of the leukemia by day 8. All CAR-treated mice were long-term leukemia-free survivors, whereas all mock-treated mice succumbed to leukemia.
Bioluminescent imaging results. NSG-immunodeficient mice were inoculated on day 0 with 5 × 105 NALM-6-GL cells and then, on day 3, received 1.5 × 107 CAR-modified T cells as designated. Mock recipients received 1.5 × 107 total cells. Transduction efficiency was ∼ 40% in both CAR groups. Mock-transduced cells were activated and expanded but were not exposed to viral vector. Bioluminescent imaging using standard techniques at the designated time points demonstrates clearance of the leukemia by day 8. All CAR-treated mice were long-term leukemia-free survivors, whereas all mock-treated mice succumbed to leukemia.
Another potentially important feature of CAR target selection is consideration of the functional significance of the surface protein for survival of the malignant cell. CD19 and CD22 are important components of the BCR complex and there are emerging data showing that CD19 contributes to oncogenesis as a downstream mediator of PAX5, which augments MYC expression.32 Nevertheless, antigen loss has been observed in the context of bispecific anti-CD19–directed therapies for B-ALL11 and after CAR therapy.28 Therefore, it is likely that alternative and, potentially multiple simultaneous, targets will be needed. CD22 and its closely related family member Siglec-G appear to play a regulatory role by inhibiting BCR signaling. Thus far, loss of CD22 expression has not been reported and further studies are needed to determine whether CD22 signaling confers any advantage to B-ALL. A variety of other B-lineage–restricted targets that may also be suitable for CAR-based therapy include CD79a, CD79b, and TSLPR. Recently, an algorithm has been developed that combines gene expression data with predicted cell surface expression to identify other potential targets.33 If we are to maximally leverage the repertoire of therapeutics available to target cell surface antigens on cancers, then further studies of this type are needed to systematically identify surface targets on leukemia cells and other malignancies.
Viral vector and transduced cell subsets
The majority of current trials of CAR-based therapies use viral-based vectors to achieve a high efficiency of gene transfer into and high level of expression on T cells (Table 1). Both lentiviral-based and non-lentiviral-based retroviral vectors are being used and there are no definitive data on which system is optimal. In all protocols, T cells are expanded in vitro, typically using mAbs (alone or in the context of beads) and cytokines (such as IL-2), both to increase cell numbers and to improve the efficiency of transduction. Genetic manipulation of T cells using nonviral systems such as transposons/transposes is also currently in clinical trials. Recent preclinical studies have suggested that transduction of specific T-cell subsets may improve the efficacy of CAR-based therapies, leading some investigators to isolate central memory T cells34 or naive T cells35 for transduction in clinical trials. An additional consideration is whether the native TCR specificity of the modified T cells will affect efficacy. To address this, some investigators are using T cells with inherent viral specificity as a source for CAR modification to potentially improve expansion/persistence to allow an “off-the-shelf” approach and/or to diminish the risk for alloreactivity when donor-derived CAR-modified T cells are administered after allo-HSCT to prevent or treat leukemia relapse.
Results of ongoing clinical trials
The initial clinical experience with CAR-modified T cells in patients with hematologic malignancies was in chronic leukemias and lymphomas. Some dramatic responses were observed using T cells modified to express a CD19-targeted CAR, including long-term remissions.36-38 The published experience in acute leukemia is more limited but is rapidly growing, with multiple clinical trials currently open. The first reported case of B-ALL treated with CAR T cells was in the context of a clinical trial at Memorial Sloan Kettering Cancer Center (MSKCC) that predominantly included patients with lymphoma. This patient was treated in second remission with T cells modified to express a CD19-targeted CAR using a γ-retrovirus after cyclophosphamide conditioning resulting in profound B-cell aplasia.39 Allo-HSCT was performed 8 weeks after CAR therapy. The MSKCC team subsequently published a series of 5 ALL patients (including the patient included in the first report) treated in the same manner,40 including 2 with > 50% blasts in the BM at time of enrollment. All 5 patients achieved complete remission to the CAR T-cell protocol with absence of minimal residual disease (MRD). Four patients went on to receive an allo-HSCT with 3 remaining in remission and 1 patient dying from posttransplant complications. One patient did not go on to allo-HSCT and relapsed 13 weeks after CAR treatment with CD19+ ALL that retained sensitivity to CAR T cells in vitro. Two pediatric patients with refractory ALL treated with CD19 CAR have also been published recently from the University of Pennsylvania.28 CAR T-cell doses were similar to MSKCC series, but the CAR construct contained CD137 (41BB) and a different anti-CD19 scFv was used. The first patient achieved an MRD-negative complete remission that has been maintained for > 1 year, whereas the second patient (who had previously been treated with CD19-directed therapy comprising blinatumomab) had a transient response but relapsed after 2 months with CD19-negative disease. The National Cancer Institute (NCI) also reported significant antileukemia effects in children with refractory ALL using a CD19-CAR containing the CD28-costimulatory domain.41 Therefore, 3 separate clinical groups have observed impressive antileukemia effects using 3 different CD19-CAR constructs in patients with refractory B-cell ALL.
One important aspect of CAR therapy is that, unlike antibody-based regimens, which mediate antitumor effects for only as long as the antibody remains present in the host, CD19-CAR T cells undergo dramatic expansion after infusion in response to CD19 antigen expressed on malignant and nonmalignant cells. Genetically modified CAR-expressing T cells can also persist for several months or even years.42 Therefore, CAR T cells represent a dynamic therapy, which is well illustrated by the fact that both response and toxicity are often delayed for several days after cell infusion.28 Finally, CAR T cells traffic to multiple tissue sites, including the CSF,28,41 an important consideration in B-ALL, in which CNS relapse is a substantial risk.
Toxicity and challenges
In the majority of the published reports in which patients with B-ALL were treated with CAR T cells, inflammatory toxicities have been observed, particularly in patients with high disease burden at the time of infusion. Furthermore, in both acute and chronic and lymphoid malignancies, inflammatory toxicity appears to correlate with disease response. The toxicities vary but typically include fever and constitutional symptoms, with hypotension and vascular leak in more severe cases. In some patients, the toxicities may be life threatening and some reports suggest amelioration of symptomatology after treatment with corticosteroids. Analysis of serum cytokines during the inflammatory syndrome shows elevation of several cytokines, including IL-6, TNFα, GM-CSF, and IFN-γ.28,38,40 One case report suggested that neutralizing antibody to soluble IL-6R may mitigate this inflammatory syndrome,28 although it is too early to know whether such treatment also limits the efficacy of the therapy. CNS toxicities have been observed with bispecific antibodies targeting CD19 and CD3, and limited clinical experience thus far is consistent with a significant incidence of reversible CNS toxicity in some patients treated with CD19-CAR therapy. More experience is needed to understand the full significance of this adverse event.
One potential long-term toxicity of CD19-CAR therapy is chronic B-cell aplasia, especially when the vectors used lead to long-term persistence, and prolonged B-cell aplasia is likely to be associated with antitumor response. It remains possible that leukemia eradication could be accomplished without prolonged persistence of CAR T cells, in which case B-cell recovery is likely. Indeed, early results from some clinical groups suggest that potent antitumor effects can occur with CD19-based therapies followed by B-cell recovery. Nonetheless, in case of long-term CD19-CAR persistence with chronic B-cell depletion, patients will need support via Ig replacement. In many ways, this condition is analogous to treatment of patients with genetic defects in B-cell developmental pathways, such as X-linked agammaglobulinemia or common variable immunodeficiency, for whom lifelong antibody replacement is required, successful, and now relatively easy to administer as a subcutaneous injection. Strategies to eradicate CAR-expressing T cells using suicide vectors are being tested as one approach to preventing such long-term toxicity, but no results are available thus far using this approach. Suicide vectors have also been proposed as a means to prevent acute toxicity such as cytokine release syndrome, although it remains unclear whether one can retain potent antitumor effects if the cells are induced to undergo apoptosis early after administration.
One of the primary challenges in treating acute leukemia compared with chronic leukemias and lymphomas is the rapid pace of ALL progression, particularly when patients are treated with large disease burdens. The achievement of complete responses in such patients, as discussed in the previous section, suggests that large tumor burdens are not a barrier to effective therapy when highly active CAR T cells are used in adequate doses. Nonetheless, if the acute inflammatory toxicity associated with CD19-CAR therapy can be prevented by treating patients with lower disease burdens, one could consider incorporating such therapy earlier in the course of disease, at which time minimal disease could be eradicated with limited inflammatory toxicity. An additional potential issue that has been suggested by some of the experiences thus far is that the collection, activation, and transduction of T cells may be more challenging in patients with B-ALL due to the potently immunosuppressive properties of ALL regimens currently in use. Indeed, transduction efficiencies appear to be lower in T cells collected from B-ALL patients compared with those collected from chronic lymphocytic leukemia patients using the same expansion process and vectors. One could potentially address this issue by harvesting T cells earlier in the disease process and cryopreserving them for potential use in CAR-based adoptive T-cell therapy at a later time if clinically appropriate. Another option is to optimize ex vivo expansion protocols for heavily pretreated patients. Finally, it remains to be seen whether antileukemic effects induce by CAR therapy in B-ALL can translate into long-term cure and potentially even preclude the need for additional consolidative therapy such as allo-HSCT. This is a central issue to consider in ALL, for which allo-HSCT has clearly been established as a potential curative option for patients able who achieve an MRD-negative remission, who have adequate organ function, and for whom an acceptable donor is available. At the same time, allo-HSCT has substantial short- and long-term toxicities and optimizing CAR-based therapy to a point where it could abrogate the need for allo-HSCT would represent a true advance in the treatment of ALL. Future studies will no doubt seek to combine CAR-based therapies, both for acute leukemia and solid tumors, with other immunomodulators such as checkpoint inhibitors as a means to further augment the potency of this new class of therapeutics.
Disclosures
Conflict-of-interest disclosure: C.L.M. holds patents with or receives royalties from the NCI. T.J.F. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Crystal Mackall, Center for Cancer Research, NCI, Building 10 - Hatfield CRC, Room 1W-3750, Bethesda, MD 20892; Phone: 301-402-5940; Fax: 301-451-7010; e-mail: mackallc@mail.nih.gov.