Abstract
Recurrent and unpredictable episodes of vaso-occlusion are the hallmark of sickle cell disease. Symptomatic management and prevention of these events using the fetal hemoglobin–reactivating agent hydroxyurea are currently the mainstay of treatment. Discoveries over the past 2 decades have highlighted the important contributions of various cellular and soluble participants in the vaso-occlusive cascade. The role of these elements and the opportunities for therapeutic intervention are summarized in this review.
Introduction
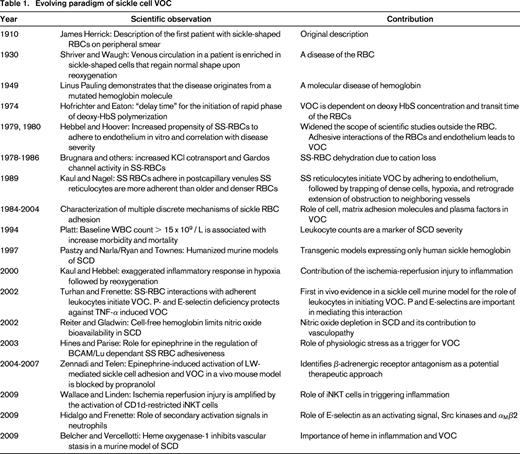
Sickle cell disease (SCD) results from a single amino acid substitution in the gene encoding the β-globin subunit. Polymerization of deoxygenated sickle hemoglobin leads to decreased deformability of red blood cells (RBCs). Through a complex interplay of adhesive events among blood cells, these altered erythrocytes can obstruct the vasculature, producing episodes of pain, hemolytic anemia, organ injury, and early mortality. Although the molecular basis of SCD is well characterized, the complex mechanisms underlying vaso-occlusion (VOC) have not been fully elucidated. Early studies using in vitro adhesion assays or a rat mesocecum ex vivo perfusion model uncovered the role of sickle RBCs (SS-RBCs) in initiating and propagating the VOC events via adhesive interactions with the endothelium.1 Preferential adhesion of low-density SS-RBCs and reticulocytes in immediate postcapillary venules leads to trapping of the older, more dense, and misshapen SS-RBCs, resulting in reduced blood flow. Random precapillary obstruction by a small number of dense SS-RBCs also contributes to VOC.1 More recent data indicate that other blood cell elements that are not directly affected by the sickle cell mutation play a direct role in VOC. A new model has been proposed in which the process is viewed as multistep and multicellular cascade driven by inflammatory stimuli and the adherence of leukocytes. Table 1 provides a summary of the evolving paradigm of sickle cell VOC.
SS-RBCs are prone to adhere
Sickle hemoglobin can cause damage to the RBC membrane from deformation by polymer formation, In addition, the mutated globin can undergo autooxidation and precipitate on the inner surface of the RBC membrane, causing membrane damage via iron-mediated generation of oxidants.2 Among the many changes that result from the damage to the SS-RBC membrane is their propensity to adhere.1,3 Some of the adhesion molecules on the surface of the SS-RBCs (eg, α4β1) have been reported to interact directly with the endothelial cell membrane (eg, VCAM-1) without the participation of an intervening plasma protein. Other adhesive interactions require a soluble bridge molecule (eg, thrombospondin, VWF). SS-RBC adhesion molecules (eg, BCAM/LU, α4β1) have also been reported to interact with the subendothelial matrix proteins (eg, laminin, VWF). SS-RBC interactions with the vascular endothelium may lead to the production of oxygen radicals by the endothelial cell and oxidant-dependent activation of the transcription factor NF-κB. NF-κB up-regulates the transcription of various genes, including adhesion molecules such as E-selectin, VCAM-1, and ICAM-1 on the surface of the endothelium. Circulating endothelial cells characterized by an activated phenotype (expression of VCAM-1 and E-selectin) and increased levels of plasma sVCAM-1 have also been reported and are reflective of continuous endothelial activation.1,4 Both endothelial selectins, P-selectin and E-selectin, have been suggested to participate in VOC.5,6 An anti–P-selectin aptamer in SCD mice resulted in a decreased adhesion of SS-RBCs, increased microvascular flow velocities, and reduced adhesion of the leukocyte to the endothelium, underscoring the importance of P-selectin as a potential therapeutic target.5 The blood group glycoprotein LW (also called ICAM-4) is an RBC adhesion receptor that can be activated by epinephrine to mediate SS-RBC adhesion to endothelial αvβ3 integrin.1 In a sickle cell murine model, these interactions led to VOC and also increased leukocyte adhesion to endothelium.7 Propranolol (a β-adrenergic receptor antagonist) and recombinant LW infusions were shown to inhibit VOC, supporting the events noted in patients who report a painful crisis precipitated by emotional stress or physical exertion.7,8 Interestingly, signals from the sympathetic nervous system transmitted by β-adrenergic receptors can also mediate circadian oscillations of leukocyte recruitment in tissues that affect the inflammatory response.9 Adrenergic control of leukocyte trafficking produces higher densities of adherent leukocytes in venules at nighttime in mice. SCD mice indeed exhibit a more dramatic VOC phenotype when the experiment is carried out at nighttime.9 Diurnal differences of admission to the hospital of SCD patients in VOC crisis have been reported, although the gradual onset of VOC crisis renders circadian influences difficult to discern.10
SS-RBCs promote inflammation
The damaged SS-RBCs and activated endothelial cells can produce a proinflammatory environment that is exacerbated during episodes of crisis. Circulating leukocytes and platelets also have an activated phenotype. Ischemia-reperfusion injury, release of free hemoglobin and heme secondary to RBC lysis, and increased production of placental growth factor (PlGF) all may contribute to the inflammatory vasculopathy.4 Levels of PlGF, an angiogenic growth factor produced by erythroblasts, are elevated in the plasma of individuals with SCD.11 Monocytes from SCD patients are activated in response to PlGF, secreting increased levels of TNF-α, IL-1, and other chemokines.11
Ischemia-reperfusion injury secondary to ongoing, intermittent microvascular occlusions promotes chronic inflammation in SCD by increased oxidant production and increased adhesion of leukocytes to the endothelium, followed by extravasation into the tissues and, finally, damage to the tissues.4 The inflammatory cascade from ischemia-reperfusion is amplified by the activation of CD1d-restricted invariant natural killer T (iNKT) cells. Compared with controls, SCD mice have more numerous and activated (CD69+, IFN-γ+) lung, liver, and spleen iNKT cells that are hyperresponsive to hypoxia/reoxygenation.12 SCD mice have increased pulmonary levels of IFN-γ+, IFN-γ–inducible chemokines (CXCL9, CXCL10) and an increased number of lymphocytes expressing the chemokine receptor CXCR3. Treatment of SCD mice with anti-CD1d antibody to inhibit iNKT cell activation or treatment with CXCR3 inhibitors reverses pulmonary dysfunction.12 Like mice, patients with SCD have increased numbers of activated circulating iNKT cells expressing CXCR3, suggesting that iNKT cells may play a critical role in mediating inflammation.
Intravascular hemolysis results in release of cell free hemoglobin in the plasma, translocation of hemoglobin to the spaces between the endothelium and the smooth muscle cells, nitric oxide depletion in the plasma and subendothelial spaces, oxidative stress, and hemin release that contribute to the inflammation.4,13 Hemin binds to the transcriptional repressor Bach-1, which regulates transcription of heme-oxygenase and other antioxidant enzymes essential for the adaptive response to enhanced intracellular hemin levels.13
The chronic inflammatory milieu in SCD activates coagulation.14 Abnormal phosphatidylserine exposure on the surface of the SS-RBCs and RBC- and platelet-derived microparticles drives thrombin generation.14 Inflammation and hemolysis promotes increased expression of tissue factor, a primary activator of the extrinsic pathway of coagulation.15 Tissue factor, however, also enhances inflammation and contributes to endothelial cell injury. Reduction of plasma levels of tissue factor in a sickle cell transgenic mouse model results in decreased plasma levels of IL-6 and soluble VCAM-1.15 Circulating platelets in SCD patients are chronically activated and express higher levels of P-selectin, CD40 ligand, and the proinflammatory cytokine TNFSF14.16 Therefore, activation of coagulation is not just a secondary event contributing to increased thromboembolic events, but also drives inflammation and vascular injury.16 An interplay of all of these processes perpetuates and feeds forward an inflammatory priming that, in the presence of a “second hit” or precipitating event, can lead to VOC.
Neutrophil adhesion and activation: critical steps for VOC
Various studies have noted a surprising pivotal role of neutrophils in a disease caused by a mutation only expressed in the erythroid lineage. Hofstra et al first reported that neutrophils could bind to SS-RBCs in vitro.17 In contrast to the previously described interaction with the endothelium, the dense cell fractions that included irreversibly sickled RBCs appeared to be the most adherent to neutrophils. In vivo evidence for this phenomenon was first reported using SCD mice that exclusively express human sickle hemoglobin. To observe dynamic interactions between the vascular wall and flowing blood cells, the cremasteric microvasculature was examined using intravital microscopy6 In this model, more striking than the occasional direct interactions of the SS-RBC with the endothelium were the prominent interactions of circulating SS-RBCs with adherent leukocytes. These interactions were increased or sustained after exogenous administration of TNF-α and frequently resulted in complete VOC. Mice deficient in both P-selectin and E-selectin were unable to recruit leukocytes at the vessel wall and were protected from TNF-α–induced VOC. This suggested that the recruitment of the adherent leukocytes to activated endothelium was necessary for the VOC process.
Heterotypic interactions are regulated by E-selectin and mediated by αMβ2 integrin
Although selectins are best known to mediate rolling interactions, allowing leukocyte recruitment to the vessel wall, they are also bona fide signaling molecules that can contribute to cell activation beyond the initial events, allowing leukocytes to firmly adhere. Although both P-selectin and E-selectin are important for adhesion of leukocytes to the endothelium, E-selectin on the endothelium appears to be crucial for generating a secondary wave of activating signals, which produces a polarized expression of activated αMβ2 integrin (also known as CD11b/CD18 or Mac-1) at the leading edge of the crawling neutrophil, mediating the capture of erythrocytes.18 Inhibition of Src family kinases, but not p38 MAPK or spleen tyrosine kinase (Syk), can reduce RBC capture. Mice deficient in the C3 complement protein, a ligand for Mac-1 integrin, have a partial reduction in RBC capture, suggesting the role of complement opsonization on the RBCs. Surprisingly, inhibition of E-selectin abrogates these effects, whereas inhibition of P-selectin has only a partial effect. E-selectin signaling can trigger “inside-out” αMβ2 activation at the leading edge of the neutrophils, whereas the engagement of platelets by activated αMβ2 can trigger “outside-in” signaling in neutrophils, leading to production of reactive oxygen species.19 Although no significant platelet accumulation has been observed at VOC sites in SCD mice (G. Chen and P.S.F., unpublished data, December 2011), there is indirect evidence for their role in VOC because inhibition of platelet activation both reduces the inflammatory milieu and the formation of platelet-monocyte and platelet-neutrophil aggregates.20
The generation of a novel synthetic pan-selectin inhibitor, GMI-1070, with maximal activity against E-selectin made it possible to further test this paradigm in VOC. As predicted by the model above, its administration to SCD mice resulted in decreased leukocyte adhesion, increased leukocyte rolling velocity, dramatic reduction of RBC capture by the neutrophils, and improved blood flow rates.21 GMI-1070 is being evaluated in a phase 2 clinical trial the results of which are pending at the writing of this manuscript.
IVIGs modulate neutrophil activation and vascular injury through FcγRIII and SHP-1
In SCD mice, IVIGs can reverse acute VOC crisis by inhibiting neutrophil adhesion to the endothelium and abrogating SS-RBC capture.22,23 Although the low-affinity FcγRIIB is crucial for the beneficial effect of IVIG in a murine model of immune thrombocytopenia and other models involving macrophage function,24 neutrophils predominantly express the “activating” FcγRIII receptor. Indeed, FcγRIII, but not FcγRIB, was found to mediate IVIG's inhibition of leukocyte adhesion, RBC capture, and reduced Mac-1 activity via recruitment of Src homology 2 (SH2)-containing tyrosine phosphatase-1 (SHP-1) in SCD mice.25 Ongoing phase 1/2 randomized double-blind placebo-controlled studies are evaluating the role of IVIG in acute VOC.
Intravascular hemolysis and VOC
It is possible that severe, acute, or subacute hemolysis triggers VOC by overwhelming the adaptive mechanisms. Clinical support for this concept comes, for example, from the observation that delayed hemolytic transfusion reactions can precipitate VOC events. Recent studies in a sickle cell murine model of hemolytic transfusion reaction have identified CXCL1 as a key inflammatory mediator of VOC. Exogenous administration of CXCL1 was sufficient to induce VOC and inhibition of CXCR2, the receptor for CXCL1, prevented the hemolytic transfusion reaction–induced VOC. Targeted inhibition of this pathway may represent a new therapeutic approach for VOC.26
Murine models of SCD support an important role of hemolysis in VOC as well. When infused into SCD mice, cell-free hemoglobin and its reactive ferric protoporphyrin-IX group hemin produce vascular stasis.27 This can be inhibited by haptoglobin or hemopexin supplementation or overexpression of heme-oxygenase-1 (HO-1).27
Proposed model of sickle cell VOC
Based on direct observations in SCD mice, the following model of VOC is proposed in which adhesive interactions of SS-RBCs and leukocytes to the endothelium play important roles in the initiation of VOC17 (Figure 1). Although the exact mechanisms remain incompletely elucidated, one can speculate that the activated adherent leukocytes, which are rigid and larger than SS-RBCs, likely drive VOC in collecting venules, whereas the SS-RBCs may contribute in smaller vessels or in situations where there is no potent inflammatory trigger. Triggers for VOC vary and can include inflammation, stress, increased viscosity, decreased flow, hemolysis, or a combination of factors as follows: (1) endothelial activation by SS-RBCs and other inflammatory mediators, (2) recruitment of adherent leukocytes, (3) activation of recruited neutrophils and of other leukocytes (eg, monocytes or iNKT cells), (4) interactions of sickle erythrocytes with adherent neutrophils, (5) vascular clogging by heterotypic cell-cell aggregates composed of SS-RBCs, adherent leukocytes and possibly platelets, (6) increased transit time to greater than the delay time for deoxygenation-induced hemoglobin polymerization, propagating retrograde VOC, and (7) ischemia as a result of the obstruction that creates a feedback loop of worsening endothelial activation. The process described above is highly dynamic and can potentially be reversed with targeted therapy.

Sickle cell VOC. SS-RBCs and other inflammatory mediators induce the activation of the endothelium. The damaged and stimulated endothelium is poised to recruit leukocytes. E-selectin on the endothelium is crucial for generating a secondary wave of activating signals, which produces a polarized expression of activated αMβ2 integrin (Mac-1) at the leading edge of the crawling neutrophil, allowing the capture of circulating discoid and sickle-shaped erythrocytes. These events culminate in VOC in the postcapillary venules.
Sickle cell VOC. SS-RBCs and other inflammatory mediators induce the activation of the endothelium. The damaged and stimulated endothelium is poised to recruit leukocytes. E-selectin on the endothelium is crucial for generating a secondary wave of activating signals, which produces a polarized expression of activated αMβ2 integrin (Mac-1) at the leading edge of the crawling neutrophil, allowing the capture of circulating discoid and sickle-shaped erythrocytes. These events culminate in VOC in the postcapillary venules.
Emerging therapies
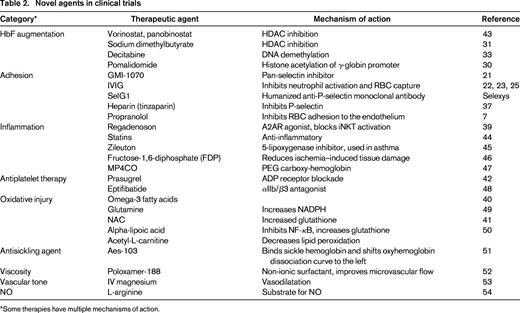
Because polymerization of the abnormal hemoglobin is a key step in the cascade of events described above, it has been an important therapeutic target for more than 5 decades. Although fetal hemoglobin (HbF)–inducing agents have had a tremendous impact in decreasing the number and severity of VOC episodes, our current understanding provides a compelling argument for clinical trials targeting other cell types and inflammatory processes. Table 2 summarizes novel agents currently in clinical trials.
HbF-inducing agents
Hydroxyurea.
Hydroxyurea is a potent inducer of HbF, and evidence over the past 25 years has documented its laboratory and clinical efficacy for both adults and children with SCD.28 Given its indisputable efficacy in responders, current efforts are directed toward expanding its utilization in both developed and developing nations. Its role as potential standard of care for all patients starting at a young age will become clearer as more data emerge on prevention of organ damage and genotoxicity with long-term use. Although hydroxyurea nonresponders are rare in childhood, approximately one-third of adults will not respond, making alternative and combination therapies a worthwhile endeavor. A recent report of coadministration of arginine as an NO substrate in a small group of patients in a randomized trial29 demonstrated an augmented HbF response and highlighted the need for future multiagent trials. A dose-limiting toxicity for hydoxyurea can be myelosuppression. Two novel HbF-inducing agents, sodium dimethylbutyrate and pomalidomide, are not myelosuppressive and actually stimulate erythropoiesis. Unfortunately, combination therapy of hydroxyurea with sodium dimethylbutyrate in a clinical trial and with pomalidomide in a murine model resulted in a blunted HbF response.30,31 Both agents are being evaluated in clinical trials individually. Hydroxyurea has multiple mechanisms by which it produces its clinical response.28 Short-term administration of either hydroxyurea or phosphodiesterase inhibitor 9 (PDE9) ameliorated VOC in a SCD murine model via cGMP-elevating effects, suggesting that hydroxyurea may have immediate benefit in the setting of VOC.32 PDE9 is highly expressed in hematopoietic cells and is thus a promising therapeutic target.
Decitabine.
Decitabine is a potent HbF inducer that acts via hypomethylation of the γ-globin promoter. In small studies thus far, it has produced a 100% response rate, even in patients who previously did not respond to hydroxyurea.33 Ongoing trials will further clarify its efficacy and tolerability. A phase 1 combination study of oral decitabine with tetrahydrouridine, a competitive inhibitor of cytidine deaminase, is targeted at improved oral bioavailability of decitabine.34
Sodium dimethylbutyrate.
Oral sodium dimethylbutyrate is currently being evaluated in clinical trials.31 Robust HbF responses were documented to IV infusions of arginine butyrate, but its short half-life makes it unattractive as a long-term therapy.
Targeting adhesion
As briefly mentioned above, GMI-1070 is a pan-selectin antagonist currently under investigation in clinical trials. This randomized, double-blind, placebo-controlled trial examined the efficacy, safety, and pharmacokinetics of GMI-1070 in hospitalized patients with SCD experiencing VOC. GlycoMimetics successfully enrolled 76 patients 12 to 60 years of age at 22 trial sites in the United States and Canada. Results of these studies have not yet been published in a peer-reviewed journal.
IVIG.
IVIG also inhibits leukocyte adhesion and activation (Figure 2). Its role in SCD patients with acute VOC is being evaluated in phase 1/2 studies via a dose-escalation strategy. Currently, the study is recruiting at the 800 mg/kg dose level and no significant adverse events have been noted thus far (N = 17; D.M. and P.S.F., unpublished observations, September 2013).

Activating and inhibitory arms of neutrophil activation. E-selectin–mediated adhesion activates Src kinases, which then leads to the up-regulation of the leukocyte integrin Mac-1 at the leading edge of crawling neutrophils. GMI-1070 works principally by inhibiting E-selectin–mediated activation. Polarized expression of activated Mac-1 allows the capture of erythrocytes. IVIG administration works by binding to FcγRIII expressed on neutrophils, leading to the recruitment of SHP-1 that inhibits Mac-1 activation.
Activating and inhibitory arms of neutrophil activation. E-selectin–mediated adhesion activates Src kinases, which then leads to the up-regulation of the leukocyte integrin Mac-1 at the leading edge of crawling neutrophils. GMI-1070 works principally by inhibiting E-selectin–mediated activation. Polarized expression of activated Mac-1 allows the capture of erythrocytes. IVIG administration works by binding to FcγRIII expressed on neutrophils, leading to the recruitment of SHP-1 that inhibits Mac-1 activation.
Tinzaparin.
Tinzaparin, a low-molecular-weight heparin, was studied in a randomized, double-blind clinical trial. Heparins act via inhibition of P-selectin–mediated adhesion35 and other anti-inflammatory effects36 in addition to their expected anticoagulant effect. After exclusion of patients with thrombocytopenia or CNS vasculopathy, 253 subjects were enrolled, 12 years of age and older, in a study in which reduced duration of VOC and no severe bleeding complications were reported.37 These results need to be validated in a multicenter study. An oral P-selectin inhibitor with an order of magnitude greater potency than heparin demonstrated improved microvascular flow in SCD patients in a phase 1 study, but has a very short half-life.35 The efficacy of SelG1, a humanized anti-P-selectin monoclonal antibody, in preventing VOC will be evaluated in an upcoming phase 2 trial.
Propranolol.
Propranolol was administered to SCD patients in a phase 1 dose escalation study and significantly reduced epinephrine-stimulated SS-RBC adhesion in a dose-dependent manner.38 Adverse events were not severe, did not show dose dependence, and were likely unrelated to drug. These results imply that β-blockers have a potential role as antiadhesive therapy via inhibition of cAMP-mediated inhibition of SS-RBC adhesion, which did not always correlate with areas of leukocyte adhesion. A phase 2 study of propranolol in SCD is currently open.
Targeting inflammation
Regadenoson.
Regadenoson is an A2A receptor (A2AR) agonist reported in a phase 1 study in SCD patients to reduce iNKT cell activation to control levels.39 The trial was conducted at 5 participating sites in the United States. No toxicity was noted. Based on positive findings in the biological end points, a phase 2 randomized, placebo-controlled trial is currently ongoing. iNKT activation in patients during VOC (n = 6) was associated with increased phospho-NF-κB p65 and A2AR expression and also higher IFN-γ levels. Although the reduction in phospho-NF-κB p65 was to baseline levels, the reduction in A2AR expression and IFN-γ levels did not reach baseline.
Antioxidant therapy
Omega-3 fatty acids.
Omega-3 fatty acids are significantly reduced in SCD patients. A randomized, placebo-controlled, double-blind trial was conducted at a single center in Sudan. Enrolled patients (N = 140) were monitored for 1 year and omega-3 treatment reduced VOC events and transfusions.40 The therapy was well tolerated and warrants further study.
N-acetyl cysteine.
N-acetyl cysteine (NAC) inhibits dense cell formation and irreversible sickle cells in SS-RBCs in vitro and restores glutathione levels toward normal. Reactive oxygen species are also produced by SS-RBCs and neutrophils18 during VOC. A phase 2 double-blind, randomized clinical trial was completed to determine the efficacy of NAC in decreasing dense cell and irreversible sickle cell formation and VOC episodes in SCD.41 Twenty-one subjects at a single center were enrolled in 4 treatment groups. NAC inhibited dense cell formation, restored glutathione levels toward normal, and decreased VOC episodes at a well-tolerated dose of 2400 mg/d (n = 6). These results need to be validated in a larger, multicenter study.
Antiplatelet therapy
Prasugrel.
Prasugrel is a novel thienopyridine P2Y12 ADP receptor antagonist that inhibits ADP-mediated platelet activation and aggregation. Phase 2 randomized, double-blind, placebo-controlled studies to examine safety were completed in adults.42 There were no hemorrhagic events requiring medical intervention in either study arm. Mean pain rates (percentage of days with pain) and intensity in the prasugrel arm were decreased compared with placebo. However, these reductions did not reach statistical significance. Platelet surface P-selectin and plasma soluble P-selectin, both biomarkers of platelet activation, were significantly reduced in SCD patients receiving prasugrel compared with placebo. Prasugrel was well tolerated and a phase 3 trial in children is registered and will be recruiting.
Conclusion
In summary, understanding of the pathophysiology of sickle cell VOC has led to several exciting new agents that are currently being evaluated. Recruitment in clinical trials and robust end points continue to represent significant challenges for translation to the clinical setting of even single agents. Given the complexity and multiplicity of events leading to the VOC event, it is possible that a multitargeted multimodal approach will likely be required to achieve the best outcome. A rigorous attention to trial design, close collaboration between basic scientists and clinicians, and a good dose of perseverance will be important in obtaining clear answers as we move forward.
This article was selected by the Blood and Hematology 2013 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2013. This article is reprinted with permission from Blood. 2013; Volume 122.
Acknowledgments
This work was supported in part by the National Institutes of Health (grants R01 HL097819, R01 DK056638, and R01 HL116340 to P.S.F.). Many important scientific and clinical contributions in SCD are beyond the scope of this report. We apologize to authors of important contributions that could not be cited because of space limitations.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: Drugs described in research studies are not being recommended for clinical off-label use; the discussion focuses on their experimental effects on the pathophysiology of SCD.
Correspondence
Paul Frenette, Albert Einstein College of Medicine, Michael F. Price Center, 1301 Morris Park Ave, Rm 101, Bronx, NY 10461; Phone: 718-678-1255; Fax: 718-678-1018; e-mail: paul.frenette@einstein.yu.edu.