Abstract
The treatment of Hodgkin lymphoma (HL) relies on multimodality treatment with standard chemotherapy, radiation therapy, and autologous or allogeneic stem cell transplantation in cases of relapsed disease. Genomic advances in HL provided insights into deregulation of key nodal signaling pathways, including the PI3K, NF-κB, and JAK/STAT pathways, which are amenable to small-molecule targeting. Understanding how HL cells interact and depend on their microenvironment for survival signals and immune protection may uncover other such pathways. Small-molecule targeting has the potential to dramatically improve treatment outcomes, especially in patients with highly refractory disease and those with poor tolerance to existing chemotherapies. As novel therapies continue to be developed for HL, the challenge will be to address the needs of high-risk groups, reduce long-term therapy-related morbidity, position current established treatments with novel therapies, and concurrently develop biomarkers to aid in patient selection. Brentuximab vedotin, which was approved in 2011, is already shifting the treatment paradigm of HL. Undoubtedly, other novel therapeutics in the pipeline will affect positively the landscape of treatment in HL.
Introduction
Hodgkin lymphoma (HL) is an uncommon malignancy with a bimodal incidence curve and approximately 9000 cases diagnosed annually in the United States. Although HL has remained a largely curable disease, approximately 20% of patients will not be cured with currently available therapy and will require subsequent treatments.
Classical HL (cHL) makes up 95% of the cases of HL and is characterized by rare Hodgkin and Reed Sternberg (HRS) cells resting in a reactive infiltrate composed of lymphocytes, histiocytes, eosinophils, and plasma cells, with HRS cells comprising < 5% of the tumor volume. The remaining cells are benign, reactive, inflammatory cells that contribute to HRS cell growth and survival. Novel therapeutics for HL should capitalize on this unique pathology and on the differential expression of cell surface antigens, oncogenic dependence of HRS cells on activated intracellular signaling pathways, and induction of anti-HRS cell immunity by modulating the microenvironment (Figure 1). In this review, we summarize recent updates on the use of monoclonal antibodies, small-molecule signal transduction inhibitors, epigenetic modulators, and agents that modulate the microenvironment.

Monoclonal antibodies
Therapeutic antibodies targeting cell surface receptors have contributed to standard-of-care treatments for multiple solid and hematologic malignancies. In HL, targeting cell surface receptors within the TNF and BCR pathways has demonstrated efficacy.
CD30
CD30 is a member of the TNF cell receptor superfamily that is highly expressed in HRS cells, but with highly restricted expression in normal cells. After several failed attempts to develop naked anti-CD30 antibody therapy, remarkable progress was achieved by conjugating the naked antibody SGN30 to antitubulin monomethyl auristatin E to generate the antibody drug-conjugate brentuximab vedotin. In a pivotal phase 2 clinical trial in patients who were previously treated with autologous stem cell transplantation, brentuximab vedotin showed an overall response rate (ORR) of 75%, with 34% of patients achieving complete remission (CR; Figure 2, Table 2).1 Similarly, brentuximab vedotin resulted in an ORR of 71% and a CR of 34% in 14 transplantation-naïve patients, resulting in a 56% conversion rate to transplantation eligibility after therapy.2 Furthermore, retreatment with brentuximab vedotin elicited a clinically relevant response in both HL and systemic anaplastic large cell lymphoma patients.3

The success of brentuximab vedotin prompts the integration of this fairly well tolerated therapy with existing standard chemotherapy. Initial experience of combining brentuximab vedotin with ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) showed significant pulmonary toxicity. When bleomycin was eliminated from this regimen, no pulmonary toxicity was observed, while maintaining a high response rate.1 Based on this data, an international randomized study comparing standard ABVD with AVD (doxorubicin, vinblastine, dacarbazine) plus brentuximab vedotin is currently being conducted. In the second-line setting, a sequential therapy of brentuximab vedotin followed by ICE (ifosfamide, carboplatin, etoposide) chemotherapy is currently being investigated in transplantation-eligible patients with relapsed and refractory HL.
Although brentuximab vedotin has significant activity in HL, other strategies to target CD30 have been disappointing. Most recently, a novel tetravalent bispecific antibody against CD30 and CD16 (AFM13), which recruits natural killer cells and macrophages to CD30+ regions to illicit an immune response, was evaluated.4 Early results from the phase 1 trial demonstrated reasonable safety but limited clinical efficacy.4
CD20
The rationale for targeting CD20 in patients with classical HL is to target HRS cells, which sometimes express CD20, but also to eliminate CD20-expressing reactive B cells in the microenvironment, which can provide survival signals to HRS cells. Two phase 2 trials of rituximab plus ABVD (R-ABVD) were reported recently. In the first study, R-ABVD therapy resulted in a 5-year event-free survival rate of 83%5 ; in the second, it resulted in a 3-year event-free survival rate of 83%.6 These trials provided rationale to conduct multicenter, randomized clinical trials comparing rituximab-ABVD with standard ABVD in patients with high-risk, advanced-stage cHL. However, with the approval of brentuximab vedotin, it remains unclear where CD20-targeted therapy would fit in the treatment of patients with classical HL.
CD40 and CD80
CD40 and CD80 were recently targeted with HCD122 and galiximab, with modest clinical activity. HCD122 (lucatumumab) is an antagonist monoclonal antibody against the transmembrane CD40 receptor. In a phase 1a clinical trial, lucatumumab administered every 4 weeks showed a 10% partial remission (PR) rate. In a subsequent phase 2 trial, 3 of 18 (17%) patients with relapsed HL achieved PRs.7 Galiximab is a chimeric anti-CD80 antibody that has also demonstrated limited clinical activity, with an ORR of 10.3% and a median PFS of 1.6 months in a panel of 30 patients with heavily pretreated cHL.8 Accordingly, no further development of these agents are being pursued in patients with HL.
PD1/PD-L1
HRS cells aberrantly express PD-L1, which is typically expressed on APCs. PDL-1 interacts with the PD-1 receptor on T cells to dampen the immune response by inhibiting TCR signaling.9 Several antibodies targeting PD-1 are currently being evaluated for the treatment of a variety of human cancers and are showing promising clinical activity. CT-011 is a humanized anti-PD-1 monoclonal antibody that was evaluated recently in patients with relapsed B-cell malignancies, but has not been investigated in patients with HL.10,11 Nivolumab (BMS-936558) is a monoclonal anti-PD-1 antibody actively being studied in a phase 1 trial for hematologic malignancies, including patients with relapsed HL. The results of this trial are highly anticipated.
Targeting oncogenic signaling pathways
Constitutive activation of the JAK/STAT, NF-κB, PI3K, and MEK/ERK pathways protect HRS cells from apoptotic signals.12 These same pathways are important in supporting the inflammatory microenvironment that contributes to survival and immune recognition of the tumorigenic HRS cells. As a result, targeting these nodal pathways by small-molecule inhibitors has a dual effect on the HRS cells and the microenvironment (Figure 1).
PI3K/AKT/mTOR
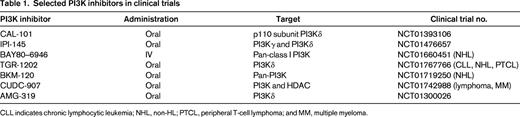
PI3K is a ubiquitously expressed phosphokinase that transduces extracellular signals from cell surface receptors such as CD40, chemokine receptors, and BCR (Figure 1). There are 4 isoforms of PI3K: α, β, γ, and δ. Activation of PI3K results in activation of downstream kinases, including AKT and mammalian target of rapamycin (mTOR), which regulate several key cellular functions including survival, metabolism, and immunity. PI3Kδ plays a key role in promoting B-cell survival and therefore is being actively targeted in B-cell malignancies. Idelalisib (GS-1101 or CAL101) is an oral PI3Kδ-selective small-molecule inhibitor that demonstrated promising clinical activity in a variety of B-cell malignancies.13 Its activity in patients with relapsed HL is currently being evaluated. In a different strategy, a dual inhibitor of PI3Kδ and PI3Kγ and PI3Kδ was developed to produce the small-molecule inhibitor IPI-145. Early results from an ongoing phase 1 clinical trial demonstrated that IPI-145 may have clinical activity in patients with relapsed HL. Other PI3K inhibitors are also being evaluated in this disease (Table 1).
To date, no data exist on targeting AKT in patients with HL. However, there are clinical data on targeting the downstream mTOR kinase. In a phase 2 study, the mTOR inhibitor everolimus produced an ORR of 47% in 19 patients (Table 2).14 A confirmatory multicenter phase 2 study that enrolled 57 patients with cHL also resulted in an ORR of 42%. Class-related side effects were observed and included stomatitis, thrombocytopenia, fatigue, and grade 1/2 pneumonitis in 6 patients.15 The results were encouraging enough to prompt additional evaluation of rapalogs in combination with histone deacetylase (HDAC) inhibitors because in vitro data suggested synergy.16,17 An ongoing phase 1/2 study combined everolimus (10 mg orally) and panobinostat (20 mg IV 3 times weekly) in patients with lymphoma. The dose-limiting toxicity was thrombocytopenia. Early results from the phase 1 portion of the study involving 13 patients with HL showed an ORR of 50%, with 71% of the patients having a tumor reduction of −12% to −72%.18 These results compare favorably with the single-agent activity of each drug (Figure 2).
JAK/STAT
The JAK/STAT signaling pathway is frequently activated in HRS cells. Activation mutations of JAK2 are rare in patients with lymphoma. In HL, the JAK/STAT pathway is activated as a result of genomic amplification of JAK2 and/or inactivating mutations in an inhibitor of JAK activity, SOCS1.12 A proof of principle of the therapeutic potential of JAK inhibitors in HL was shown by a phase 1 study of the JAK inhibitor SB1518, a selective inhibitor of JAK2 and FLT3. In that study, 14 of 34 total lymphoma patients had cHL. Of these 14 patients with cHL, 6 patients had stable disease with treatment.19 In addition to a direct antitumor effect, JAK inhibitors may also induce favorable immunomodulatory effects. AZD1480, a JAK1/2 inhibitor, displayed immunomodulatory effects at low concentrations by down-regulating the expression of Th2 cytokines and chemokines (IL-13 and TARC), as well as STAT3-mediated reduced expression of PD-L1 and PD-L2, which are involved in immune recognition. These results support the theory that JAK signaling modulates the microenvironment of HL and controls autoimmunity in HL.9,20-22
NF-κB
NF-κB is activated in HRS cells by multiple genetic alterations. REL, a transcription factor of the NF-κB family, is amplified in ∼ 50% of cHL. Mutations and deletions of NF-κB inhibitors such IκBα have also been observed.12 In other cases, aberrant secretion of cytokines in HL microenvironment can also result in activation of NF-κB. Based on preclinical data demonstrating that bortezomib can inhibit NF-κB and induce cell death in HRS-derived cell lines, bortezomib was evaluated in patients with relapsed HL. However, 2 independent small studies reported that bortezomib has no significant clinical activity in patients with relapsed HL.23,24 Preclinical data suggested a synergy between bortezomib and chemotherapy, prompting combination of bortezomib with ICE chemotherapy, which showed encouraging early results: the ORR was 70% and the CR was 33%.25 As brentuximab vedotin is being combined with ICE chemotherapy in transplantation-eligible patients with relapsed HL, it is unlikely that bortezomib plus ICE will advance in the clinical development for this disease.
Epigenetic therapy
Epigenetic events such as acetylation, methylation, ubiquitination, and phosphorylation of histones control the accessibility of the chromatin structure for DNA transcription, replication, repair and cellular development. Human HDACs are classified into 4 classes: class I includes HDAC 1, 2, 3, and 8, which are localized to the nucleus with ubiquitous tissue expression; class II includes HDAC 4, 5, 6, 7, 9, 10, which have variable cellular localization; class III includes NAD-dependent homologs of yeast, SIRT 1-7, which are not being targeted by currently available HDAC inhibitors; and class IV is composed of HDAC 11.26 Preclinical and clinical experiments have demonstrated that HDAC inhibitors may have a potential therapeutic value in patients with HL by a direct antitumor effect plus an indirect immunoregulatory effect (Figure 3).16 HDAC inhibitors reduce the secretion of the chemokine TARC (CCL17), which is responsible for T-cell chemotaxis.16,27 HDAC inhibitors may also restore antitumor immunity by up-regulating OX40L and disrupting the interaction between PD-1 and PD-L1/2 interactions, which are required for the generation of memory T cells and self-recognition by regulatory T cells.28

Base on these preclinical data, several HDAC inhibitors have been evaluated recently for the treatment of relapsed HL and have shown encouraging results (Figure 1). Vorinostat is one of the earliest HDAC inhibitors with activity against class I and II HDACs. In vitro experiments demonstrated that vorinostat has a higher IC50 against HL cell lines compared with other HDAC inhibitors. This weak in vitro activity was also associated with a weak clinical activity, with only one patient achieving PR in a phase 2 trial involving 25 patients with HL (Table 2).29
Mocetinostat
Mocetinostat (MGCD0103) is an oral nonhydroxamate HDAC inhibitor that selectively inhibits HDAC 1, 2, 3 and 11.30,31 A phase 2 trial of 51 patients with relapsed or refractory HL evaluated dose escalation of mocetinostat with a cohort of 23 patients treated with 110 mg of mocetinostat 3 times a week and a second cohort of 28 patients treated with 85 mg of mocetinostat 3 times a week.32 Toxicities include thrombocytopenia, fatigue, pneumonia, anemia, and pericardial effusion, but the ORR was 27%, which is suggestive of clinical activity (Figure 2, Table 2).32
Panobinostat
Panobinostat is an oral pan-HDAC inhibitor. A phase 2 clinical trial testing 40 mg panobinostat in 129 patients with relapsed cHL showed an ORR of 27%, with 5 CRs and 30 PRs (Figure 2, Table 2).33 Biological activity was detected by an early decline in serum TARC levels. Based on preclinical data suggesting synergy with mTOR inhibitors, panobinostat in combination with everolimus is being studied currently.16,17
Entinostat
Entinostat is a class I isoform-selective HDAC inhibitor shown preclinically to induce apoptosis, block promoting factors, and up-regulate T-cell coactivators.34 These results led to the phase 2 study administering entinostat in 2 dosing schemas: 10-15 mg every other week (n = 33) or 15 mg weekly for 3 weeks (n = 16) in 28-day cycles.35 Thirty-eight patients were evaluable for response, resulting in 6 patients with PR (ORR 16%) and 45% with SD (Table 2).35 Progression-free survival was short at 3.8 months.35
Immunomodulators
The importance of cellular immunity in regulation of HL suggests that immunomodulatory agents may have activity in this disease. Lenalidomide is an immunomodulatory agent with antiangiogenic properties. A phase 2 multicenter study of lenalidomide given in an oral daily dose of 25 mg on days 1-21 of a 4-week cycle in patients with relapsed or refractory HL demonstrated modest single-agent activity.36 Thirty-eight patients with a median of 4 prior therapies demonstrated an ORR of 19%, 1 patient with CR, 6 patients with PR, and 5 patients with stable disease (Figure 2). The most common dose-limiting toxicities were cytopenias, rash, and hepatic toxicity.
Conclusion
The development of new therapies has been stagnant in HL for more than 30 years. Since 1977, brentuximab vedotin is the only new agent to be approved by the Food and Drug Administration for the treatment of HL. Many new small-molecule inhibitors have promising clinical activity, paving the way for a revolution in the treatment of HL (Figure 2). Although some seem to have direct activity as single agents (Table 2), others potentially reverse chemoresistance and may have activity when combined with either other small-molecule inhibitors or traditional chemotherapy. Future investigations should focus on identifying predictive biomarkers to help select patients who are more likely to respond to these novel agents.
Disclosures
Conflict-of-interest disclosure: C.L.B. declares no competing financial interests. A.Y. has received research funding from Gilead, Novartis, Curis, and Johnson and Johnson and has received honoraria from Seattle Genetics, Gilead, Novartis, and Sanofi. Off-label drug use: Emerging treatment strategies using brentuximab vedotin, PI3K inhibitors, HDAC inhibitors, and ibrutinib in patients with HL.
Correspondence
Anas Younes, Lymphoma Service, Memorial Sloan-Kettering Cancer Center, 1275 York Ave, Box 330, New York, NY 10065; Phone: 212-639-7715; Fax: 646-422-2291; e-mail: younesa@mskcc.org.