Abstract
Hereditary hemochromatosis (HH) due to homozygosity for the C282Y mutation in the HFE gene is a common inherited iron overload disorder in whites of northern European descent. Hepcidin deficiency, the hallmark of the disorder, leads to dysregulated intestinal iron absorption and progressive iron deposition in the liver, heart, skin, endocrine glands, and joints. Survival is normal if organ damage is prevented by early institution of phlebotomy therapy. HH arthropathy is the symptom most affecting quality of life and can be debilitating. Genotype screening in large population studies has shown that the clinical penetrance of C282Y homozygosity is highly variable and can be very low, with up to 50% of women and 20% of men showing a silent phenotype. Targeted population screening for the HFE C282Y mutation is not recommended at present, but might be reconsidered as a cost-effective approach to management if counseling and care were better organized and standardized. Referral of patients to the blood center for phlebotomy therapy and use of HH donor blood for transfusion standardizes treatment, minimizes treatment costs, and may benefit society as a whole. Physician practices should be amended such that HH subjects are more frequently referred to the blood center for therapy.
Introduction
Hereditary hemochromatosis (HH) is an inherited iron-overload disorder caused by excessive and dysregulated intestinal iron absorption that is mediated at the level of the duodenal enterocyte.1 It is the most common single-gene disorder in whites of northern European descent, with homozygosity for a point mutation, 845G→A in the HFE gene, resulting in the substitution of a tyrosine for a cysteine at amino acid 282 of the HFE protein (Cys282Tyr or C282Y), found in 85% to 100% of patients with the clinical phenotype.2,3 An additional variant HFE allele, His63Arg (H63D), may also contribute to iron overload in some cases.3 In this review, the terms HH and HFE-hemochromatosis refer to the autosomal-recessive disorder associated with homozygosity for the HFE C282Y allele.
In patients with HH, increased iron absorption throughout life may lead to progressive iron deposition and injury to multiple tissues and organs, with parenchymal cells of the liver, endocrine organs, heart, and joints being particularly susceptible to damage.4,5 If untreated, the disease can result in advanced fibrotic liver disease, cirrhosis, hepatocellular cancer, sexual dysfunction due to hypogonadotrophic hypogonadism, cardiomyopathy, diabetes mellitus, destructive osteoarthritis, and diffuse skin pigmentation. Fatigue, impotence or loss of libido, and arthritis are the 3 most common presenting symptoms, with elevated laboratory indices of iron stores (as reflected in the serum ferritin level) and abnormal iron absorption and recycling (as reflected in the transferrin saturation value) being the most common laboratory findings. Reactive iron species, or labile plasma iron, results when plasma transferrin levels are insufficient to bind all available absorbed and recycled iron. Excess iron damages cells in the liver and other parenchymal organs through oxidative stress and generation of reactive iron species via the Fenton reaction.6
Genetic background and molecular pathogenesis of disease
HH is also referred to as type 1 hemochromatosis to distinguish it from other forms of genetic hemochromatosis. These include rare autosomal-recessive disorders caused by mutations in the transferrin receptor 2 gene (TfR2), which results in a phenotype similar to HH, and in the hepcidin (HAMP) or hemojuvelin (HJV) genes, which result in a much more severe phenotype referred to as juvenile hemochromatosis.3 Rare autosomal-dominant forms of iron overload due to mutations in the ferroportin gene can present with a variable phenotype, depending on the nature of the variant ferroportin allele.7
HFE is an MHC class-I like gene located near the MHC locus on chromosome 6. Its evolution from MHC-like origins may explain the large number of HFE polymorphisms that have been identified, only 2 of which, C282Y and H63D, appear to be clinically significant.8 Loss of an internal disulfide bond in the HFE C282Y gene product disrupts the tertiary configuration of the HFE protein such that it undergoes intracellular degradation and is not expressed on the cell membrane. HFE plays an important role in regulating hepcidin expression in the liver, and HFE mutations result in inappropriately low hepcidin levels. Impaired hepcidin synthesis is the central pathogenic factor in HH because hepcidin, through its interaction with and suppression of ferroportin activity, is the predominant regulator of systemic iron homeostasis in humans.9 Under physiologic circumstances, plasma transferrin saturation regulates liver hepcidin expression via a signaling pathway that includes HFE, TfR2, and a bone morphogenetic protein–hemojuvelin receptor complex.10 Hepcidin is secreted into the blood, where it acts as a negative regulator of iron absorption from the gut and iron egress from macrophages through its interaction with the transmembrane cellular iron exporter ferroportin.11 Hepcidin binding induces the internalization and degradation of ferroportin. Hepcidin deficiency thus results in unregulated, excess uptake of dietary iron and increased iron release from macrophages containing phagocytosed RBCs. The exact mechanism by which HFE gene mutations mediate defective hepcidin synthesis is not known, but HFE may be involved in sensing transferrin bound iron levels through its association with transferrin receptor 1 (TfR1) on the cell surface. Transferrin binding to TfR1 may free HFE to bind to TfR2, which may activate the signaling pathway for hepcidin transcription.
Natural history, population distribution, and clinical manifestations
The availability in the late 1990s of a genetic test for the C282Y and H63D alleles of the HFE gene and its widespread use in population screening studies in the past decade have revealed that the allele frequencies for C282Y and H63D and the prevalence of C282Y homozygosity are considerably more common than expected. However, these studies also showed that only a minority of patients with HH will exhibit clinical symptoms of disease. A recent large genetic-screening study in North America and Canada (the Hemochromatosis and Iron-Overload Screening [HEIRS] study) involved genetic testing of nearly 100 000 subjects (44 000 whites) and showed that in whites the prevalence of C282Y homozygosity was 0.4% to 0.5% (1 in 200 to 1 in 250), the prevalence of C282Y/H63D compound heterozygosity was 2%, and the prevalence of H63D homozygosity was 2.4%.12 The carrier rate (simple heterozygosity) for C282Y was 10% and for H63D was 24%, such that only 61% of whites had wild-type or nonmutated HFE alleles. In contrast, the prevalence of C282Y was extremely low in nonwhites, although heterozygosity for H63D was found in 8% of Asians.
The disease penetrance of HH has been the topic of substantial national and international debate and controversy. In patients detected in the clinic and referred for medical care, the presence of disease symptoms ranges from 30% to 70%, with men developing symptoms and signs of organ impairment at a rate 5 to 10 times that of women.13-15 Referral bias may result in skewed ascertainment of symptoms, such that 30% of HH patients referred to a tertiary gastroenterology practice were documented to have cirrhosis in one study, in contrast to 2% referred to a blood center practice.16,17 The risk of clinical complications is directly associated with the amount of excess iron burden. Patients are currently detected earlier in the course of the disorder due to the widespread incorporation of iron-screening indices in routine laboratory assessments, so that cardiomyopathy and diabetes mellitus, both late-stage developments that almost always occur in association with advanced liver disease, are rarely seen. Cirrhosis, liver failure, and hepatocellular carcinoma are the major causes of death in HH, with the risk of cirrhosis 13 times higher than in the age- and sex-matched non-HH population and the risk of hepatic cancer 70 to 100 times that of the general population.18 Cirrhosis is rarely seen in patients with ferritin values < 1000 μg/L, but is significantly associated with higher ferritin values.16 Hepatic cancer generally, but not always, occurs in the setting of preceding cirrhosis. Sexual dysfunction in men is common, reported in 20% to 30% of medically referred patients, and fatigue and lethargy are reported in another 30% to 40%.4 Although these are also common symptoms in non-HH subjects, several large population-screening studies demonstrated that both impotence and fatigue were significantly more common in men with HH than in men without HFE mutations.12,19 Patients with HH have a normal life expectancy if they are treated before significant iron overload occurs and before end-organ damage to the liver and heart develops; in contrast, HH patients with cirrhosis have a decreased life expectancy compared with the general population.18
Arthritis is the symptom that most commonly affects quality of life in patients with HH.20 It is a symmetrical polyarthritis involving both large and small joints, and affects 30% to 60% of HH patients diagnosed in the clinic. A predilection for pain, redness, and swelling in the second and third metacarpophalangeal joints is unique to HH and is frequently used to discriminate HH arthritis from that in non-HH control populations. Sclerosis, spurs, joint space narrowing, and chondrocalcinosis may be seen on radiography. A particularly painful, destructive osteoarthritis and osteonecrosis are also seen in the larger weight-bearing joints of HH patients. In one prospective study, the likelihood of at least one total joint replacement (hip, knee, or ankle) was 31% by age 70 in a cohort of 244 HH subjects; in contrast, only 1% of non-HH controls followed concurrently for combinations of other variant HFE alleles underwent joint replacement.21 Ferritin value at diagnosis was the single factor most strongly associated with subsequent joint replacement, with a 5-fold increase in risk in subjects with ferritin > 1000 versus < 1000 μg/L at diagnosis. It is not known if initiation of phlebotomy early in life can prevent the debilitating osteoarthritis of HH.
The considerable variability in disease penetrance seen in subjects with HFE C282Y homozygosity can be attributed in part to non-HFE environmental and genetic influences. Environmental factors that increase the risk of advanced liver disease include coexisting nonalcoholic fatty liver disease (NAFLD), excessive alcohol consumption, viral hepatitis, and increased red meat ingestion.22 Regular blood donation or ingestion of proton pump inhibitors, which impair acidification in the proximal duodenum and retard dietary iron absorption, may delay or avert systemic iron loading. NAFLD is a particularly common process that can act as a cofactor in HH disease to elevate the serum ferritin and liver transaminases and hasten the development of liver disease. The main genetic factor that predisposes to accelerated iron loading in HH is male sex, likely related to recurrent blood loss in women due to menses and childbirth and varying dietary habits among men and women.23
The occurrence of clinical iron overload disease in patients with compound heterozygosity for C282Y/H63D is extremely rare.24 Elevations in iron indices are more common with this genotype than in the general population, but are not associated with tissue or organ damage in the absence of another process. Patients with elevated liver transaminases in the setting of C282Y/H63D nearly always have an associated primary hepatic inflammatory process, most commonly NAFLD. A small proportion of subjects with H63D homozygosity may also have elevated iron indices, but do not develop iron overload disease in the absence of another primary hepatic inflammatory process.
Diagnosis
Elevation in transferrin saturation is the first biochemical abnormality in HH, and may be present in the absence of iron overload. This is followed by elevation in serum ferritin, which reflects excess total body iron burden. The serum transferrin saturation is the single most helpful screening test for HH. It is a highly specific phenotypic marker for the presence of one or more variant HFE alleles and was shown to be elevated in 73% to 84% of men and 69% to 73% of women with HH in large population-screening studies.12,19 The definition of an elevated transferrin saturation differs by study, but is generally > 50% to 55% in men and > 45% in women. The yield of the test is increased by combining it with a ferritin level. Ferritin is elevated in 82% to 88% of men and 52% to 57% of women in screening studies; the upper limit of normal is generally 300 in men and 200 μg/L in women.12,19 However, although serum ferritin is a highly sensitive marker for HH, it has low specificity for the disorder; in the HEIRS study, 26% of all men without HFE polymorphisms had an elevated ferritin.12 Ferritin is an acute-phase reactant that is elevated in necroinflammatory liver diseases such as alcoholic liver disease, NAFLD, and viral hepatitis, as well as in nonhepatic chronic inflammatory states, including malignancy and infection. A combination of transferrin saturation and ferritin testing is generally recommended in screening of high-risk populations, including family screening, and in diagnostic testing due to symptoms. An HFE genotype should be obtained if both tests are elevated or, in screening situations, if only the transferrin saturation is elevated. Liver function tests (aspartate transaminase and ALT) are useful to quantitate the degree of liver injury, to assess the response to therapy, and to determine whether other processes in addition to HH are present.25 The complete blood count may also be useful in the diagnosis of HH. The hemoglobin and the mean corpuscular volume (MCV) are at the upper limit of the reference range, with MCV's commonly in the 95 to 99 fL range. The hemoglobin and the MCV both decline with phlebotomy therapy and may be used to guide the end points of therapy.26
Noninvasive assessment of iron burden in the liver or heart using MRI generally does not offer any clinically helpful information and is not routinely indicated. Liver biopsy is not necessary for diagnostic purposes, but may be performed for prognostic reasons in patients in whom cirrhosis is strongly suspected. Liver ultrasound should be performed every 6 months as surveillance for hepatocellular cancer in patients with biopsy-proven cirrhosis or, in the absence of a biopsy, in patients with initial ferritin levels well above 1000 μg/L. Liver ultrasound is also useful to document the presence of coexisting steatosis in patients with persistent ALT elevations after normalization of ferritin with phlebotomy.
Population screening and disease penetrance
HH is a disorder that fits the accepted criteria for population screening with a genetic test: (1) it is relatively common in the targeted (white) population; (2) a long asymptomatic period exists, allowing screening and treatment to occur before irreversible organ damage develops; (3) treatment is inexpensive and easily accessible; and (4) early intervention can prevent serious and life-threatening sequelae, making population screening a cost-effective strategy for preventing illness and death. However, a series of large population-screening studies in the past decade yielded highly variable estimates of clinical penetrance, calling into question the advisability of universal screening. In one questionnaire-based survey of 41 038 patients attending a health appraisal clinic, clinical penetrance for HFE C282Y homozygosity was estimated to be only 1%; however, a quarter of the C282Y homozygotes were excluded from the analysis due to a known diagnosis of HH, likely leading to an underestimate of clinical penetrance, and in-person clinical examinations were not performed.27 In contrast, the prevalence of at least moderately advanced fibrotic liver disease among male and female C282Y homozygotes in 65 238 Norwegian subjects was 10% and 2.4%, respectively.28 Similarly, advanced hepatic fibrosis was found in 4.3% of asymptomatic C282Y homozygotes identified through screening of 11 037 Australian subjects,29 and cirrhosis was documented in 5.6% of men and 1.9% of women in a separate cohort of 672 asymptomatic C282Y homozygotes.14 A higher daily intake of alcohol among Australian (14 g/d) compared with American (3.6 g/d) HH subjects may contribute to these differences12,19,30
An Australian screening study of 31 000 subjects demonstrated that iron-overload–related disease, as defined by objective criteria and recorded through structured interviews, was present in 28.4% of men and 1.2% of women.24 In that study, 45% of C282Y homozygous males and 8% of females had ferritin values > 1000 μg/L; arthritis of the second and third metacarpophalangeal joints was more common in C282Y homozygotes than in controls; and 9% of C282Y homozygotes developed advanced liver disease, including hepatic cancer. The Australian data were compelling enough for the investigators to launch 2 additional community screening programs, 1 recruiting subjects in the workplace and 1 targeting teenagers in high school. Subjects assessed as needing phlebotomy therapy were offered treatment through the Australian Red Cross, where their blood could be used for transfusion. The outcomes of these studies were strikingly positive, with high participation rates, high compliance with phlebotomy, decreased subject anxiety on follow-up interviews, and substantial participant satisfaction at having had the diagnosis of HH made early. The conclusion from these studies is that targeted screening of the population at highest risk of morbidity related to C282Y homozygosity (white males) using either biochemical phenotype (serum iron studies) or genotype is reasonable and cost-effective and does not result in psychologic harm, discrimination, or stigma.29 To maximize cost-effectiveness and minimize the provision of ambiguous genetic information, it has been recommended that genotypic screening probe only for the HFE C282Y allele.31
Treatment
Phlebotomy therapy has been the mainstay of treatment for HH patients for > 60 years.32 A single 500-mL unit of blood contains ∼ 220 to 240 mg of iron, depending on the donor's hemoglobin. The drop in donor hemoglobin stimulates erythropoiesis and mobilizes iron from parenchymal storage sites into the BM, where it is used to make more RBCs. The ferritin level declines with each whole blood donation; there is a weak but significant linear correlation between the initial ferritin level and the number of blood donations that are necessary to achieve the end point of therapy. The transferrin saturation does not change until late in the course of treatment; monitoring of the saturation is not indicated during therapy.
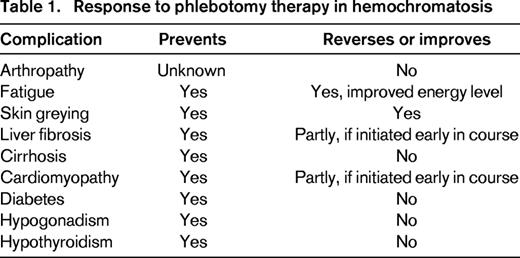
There are no randomized trials of the efficacy of phlebotomy in HH. However, serial phlebotomy therapy lowers the ferritin level and depletes the body of excess iron and, when initiated before the development of cirrhosis or diabetes, significantly reduces the morbidity and mortality of HH.18 In symptomatic patients, phlebotomy can decrease fatigue and skin pigmentation and, in some cases, reverse hepatic fibrosis.33 However, phlebotomy is ineffective in ameliorating the arthritis, hypogonadism, or cirrhosis of HH and cannot reverse the diabetes (Table 1). There is widespread consensus to initiate phlebotomy therapy in all patients with ferritin levels > 1000 μg/L because levels in this range are strongly associated with end-organ damage.34 Indications for starting therapy are less robust for HH patients with milder elevations in ferritin ranging from 200 to 600 μg/L, particularly if they have minimal or no symptoms referable to HH. Such patients may not develop progressive disease and may not experience further increases in ferritin with time. However, because phlebotomy is safe, inexpensive, accessible, and may even provide collateral benefit to others (blood donation), treatment is usually initiated. We recommend initiating phlebotomy therapy for all C282Y homozygotes with ferritin levels above the reference range.
The target of phlebotomy is a ferritin level between 50 and 100 μg/L, a value that ensures the depletion of excess iron but leaves the subject with some iron reserve. Iron depletion but not iron deficiency is the goal; iron deficiency is surprisingly easy to achieve and should be avoided. Symptomatic iron deficiency was present in 11% (10 of 90) of previously treated subjects referred to our center and was of sufficient severity to merit a short course of oral iron replacement. The pace of initial phlebotomy is dependent on the starting ferritin level (Figure 1). For a ferritin > 1000 μg/L, phlebotomy is generally initiated weekly, for levels of 500 to 999, biweekly; for levels of 200 to 499, every 4 to 8 weeks. The pace may be slowed when the ferritin approaches the targeted range of 50 to 100 μg/L. Serum ferritin is usually monitored every 2 to 4 phlebotomy sessions, depending on the pace of the treatments, until it is < 200 μg/L and then every 1 to 2 donations until it falls below 100 μg/L. The minimal hemoglobin to proceed with phlebotomy should be 12.5 g/dL. Because HH subjects typically present with hemoglobin levels in the 14 to 15 g/dL range, by the time the value has dropped to 12.5 g/dL, a substantial fraction of the circulating RBC pool has been depleted and the subject is likely to experience fatigue. It has been our experience that the majority of HH donors can successfully undergo iron depletion therapy while maintaining a minimal hemoglobin level of 12.5 g/dL.

Algorithm for management and monitoring of phlebotomy therapy in HFE C282Y homozygous HH.
Algorithm for management and monitoring of phlebotomy therapy in HFE C282Y homozygous HH.
Requirements for maintenance phlebotomy are highly variable. To avoid iron reaccumulation, we advise keeping the ferritin in the 50 to 100 μg/L range during maintenance, although others advise allowing the ferritin to rise to the upper reference range (200-300 μg/L); there are no controlled trials to support either approach. Maintenance intervals vary from every 4 weeks to once yearly and should be tailored to the individual patient. Some patients will not reaccumulate iron at all and will only need periodic monitoring.
Role of the blood center in HH management
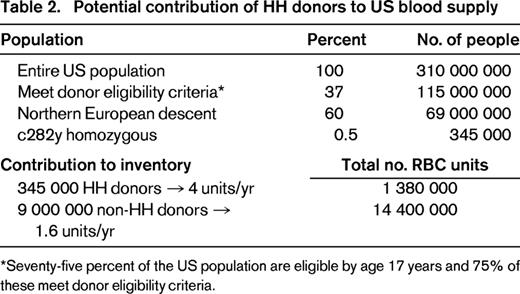
In a cohort of 130 subjects with variant HFE alleles referred to a blood center for management, 76% met eligibility criteria for allogeneic blood donation and 55% had previously been blood donors before being made aware of their diagnosis.17 HH donors were documented to keep their donation appointments more regularly than non-HH donors and to experience deferrals for low-screening hemoglobin (< 12.5 g/dL) at a significantly lower rate than non-HH donors (< 1% vs 10% of donation visits). The average untreated male with HH has approximately 8 g of excess body iron at diagnosis; at a distribution of 240 mg iron per whole blood unit, this is equivalent to 33 units of blood. Recognizing that HH subjects could constitute a safe source of blood for transfusion, the US Food and Drug Administration in 2001 issued a Guidance allowing blood centers to submit variances to federal code to allow blood from HH donors to be used for transfusion.35 To ensure adequate safeguards around this process, the variance must include provisions that: (1) the donor otherwise meets all allogeneic donor criteria, (2) phlebotomy is provided free of charge to all HH subjects who come to the blood center, (3) incentives for HH donors to be untruthful in responding to standardized health history screening questions are minimized, (4) a medical prescription for phlebotomy therapy including frequency and hemoglobin threshold is provided by the donor's physician, and (5) an abbreviated physical examination is performed at each visit if the donor donates more often than every 8 weeks. In the intervening 12 years since the Guidance was issued, 163 blood donor centers in 43 states in the United States have submitted variances and implemented polices for collection of blood from HH donors. HH subjects derive considerable satisfaction from knowing that their blood units are being used to save lives rather than being discarded.17 It is estimated that routine referral of HH subjects to blood centers for phlebotomy care could supplement the US blood supply by an additional 1.3 million RBC units per year, possibly help to avoid periodic RBC shortages, avoid wastage of safe units, and decrease the costs of care (Table 2). The percentage of therapeutic phlebotomies in HH subjects performed in a blood center versus a physician's office is not known.
Treatment of HH in the donor center may also help to ensure that the minimal acceptable hemoglobin level to proceed with a donation does not fall below 12.5 g/dL, the legal limit for blood donation. Use of the donor hemoglobin threshold as the criteria for phlebotomy may thus help to prevent the overly aggressive iron depletion that is sometimes seen in clinical practice. People with C282Y/H63D and H63D/H63D genotypes and elevated ferritin levels are also frequently referred to the blood center for treatment. These subjects in general do not have the same susceptibility to parenchymal iron overload and end organ damage as C282Y homozygotes and may not need an aggressive approach to phlebotomy therapy. Careful consideration should be given to the indications for and frequency of phlebotomy therapy in these subjects and ferritin levels should be closely monitored during their treatment course.
In smaller, hospital-based blood banks with active HH recruitment and management programs, HH donations can contribute substantially to the facility's blood supply, constituting 10% to 40% of available RBC inventory.17 In centers without active recruitment, only 0.4% of whole blood donations were derived from HH subjects.36 One additional advantage of HH treatment in the blood center is that phlebotomy may be accomplished with use of a double-RBC apheresis device.37 During double-RBC apheresis procedures, 2 packed RBC units are collected from a single donor in an isovolemic manner, with saline infusions replacing the RBC volume lost. The total iron removed is ∼ 410 mg. Donors must have a slightly higher qualifying hemoglobin of 13.3 g/dL, but this approach may reduce the frequency of visits by half while still accomplishing the same pace of iron removal.
Conclusion
Routine referral of HH subjects to the blood center for phlebotomy care could be factored into the current debate on targeted population screening for hemochromatosis. Part of the debate involves the excess costs of diagnosing, monitoring, and treating a disorder with an uncertain likelihood of causing clinical disease in the future. With routine referral to the blood center, treatment costs are minimized, phlebotomy therapy is performed by skilled staff in a highly regulated environment, and a collateral societal benefit may occur. Economic models of population screening for HH have suggested that genetic screening would be cost-effective only if at least 20% of subjects developed life-threatening conditions.38,39 Although life-threatening complications are seen in < 5% to 10% of HH subjects, debilitating processes such as small and large joint arthritis occur with a frequency of at least 20%.20 Routine referral to the blood center may encourage the organization of prospective clinical trials to determine whether targeted screening of at-risk subjects at a young age, perhaps in the late teens or early 20s, could prevent the development of some of the more debilitating and irreversible features of hemochromatosis.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Susan F. Leitman, MD, Chief, Blood Services Section, Department of Transfusion Medicine, NIH Clinical Center, Building 10, Room 1C0-711, Bethesda, MD 20892; Phone: 301-496-4506; Fax: 301-402-1360; e-mail: sleitman@nih.gov.