Abstract
Refractory aplastic anemia (AA) is defined as a lack of response to first-line immunosuppressive therapy (IST) with antithymocyte globulin and cyclosporin and is manifested as persistence of severe cytopenias at 6 months after IST. Although supportive care is critical for AA patients, it is of paramount importance for refractory disease in view of the longer duration of pancytopenia and susceptibility to life-threatening infections due to IST. Improvements in supportive care have largely contributed to better outcome over the past 2 decades, with 5-year overall survival reaching 57% during 2002 to 2008 for patients with AA unresponsive to initial IST. Exclusion of hypocellular myelodysplastic syndrome and constitutional BM failure masquerading as apparent idiopathic AA should be done in conjunction with centers of excellence. Hematopoietic stem cell transplantation is indicated if refractory AA patients are fit and have a suitably matched donor, either a sibling (> 40-50 years) or unrelated donor. Patients lacking a fully matched donor should be considered for a second course of antithymocyte globulin plus cyclosporin, although response in the refractory setting is only ∼ 30% to 35%. Response may also occur with alemtuzumab or the thrombopoietin mimetic eltrombopag in refractory AA. The emerging data for alternate donor (cord or haploidentical) transplantation in AA has provided additional therapeutic choices to consider in refractory disease.
What does refractory aplastic anemia really mean?
Refractory aplastic anemia (AA) has been defined as lack of response with persistence of severe pancytopenia at 6 months after 1 course of immunosuppressive therapy (IST).1 However, at this stage of the disease, important issues need to be considered before deciding how best to treat the patient; referral to a center with specific expertise in AA should be considered2,3 for the following reasons:
First, a careful reassessment should be made to confirm that the diagnosis is still AA and not hypocellular myelodysplastic syndrome (MDS); the morphological distinction between the 2 is not always easy and the BM aspirate and trephine should be assessed for dysplastic megakaryocytes and granulocytes, presence of abnormal localization of immature precursors (ALIPs) or blasts, abnormal sideroblasts, and increased reticulin, any of which would be in keeping with MDS.4
Second, advantage should be taken for using the latest molecular testing that may be available at a specialized center, to help exclude not only hypocellular MDS, but also constitutional BM failure. At the same time, detailed family history and meticulous clinical examination should be performed to detect subtle and more recently recognized clinical features of constitutional BM failure syndromes, which more often present with a milder phenotype in adults than in children.5
Third, AA is a rare disease, and patients should be given the opportunity to discuss fully with experts the pros and cons of the different treatment options, both transplantation and nontransplantation, that are available for refractory AA. Once reassessed, patients should be considered for enrolment into a clinical trial whenever possible, because most of the treatment options for refractory severe AA (SAA) are considered experimental.
The lack of response to a first course of antithymocyte globulin (ATG), may mean that: (1) the pathogenetic mechanism is not immune mediated and may represent constitutional AA and/or telomere disease, although sometimes response to IST occurs in dyskeratosis congenita and short telomeres do not correlate with lack of response6 ; (2) there may be extreme hematopoietic stem cell (HSC) exhaustion, making it unlikely that further IST will be effective, as implied from one study in which patients treated with a third course of ATG who had failed to respond to previous courses showed no sustained response to a third course7 ; and (3) failure to respond may be due to inadequate immunosuppression, although the use of additional immune suppressive drugs such as sirolimus or MMF to ATG and cyclosporin (CSA) does not increase the response rate. Factors predicting nonresponse to a first course of ATG include older age and low absolute reticulocyte count and absolute lymphocyte count. The combination of high absolute reticulocyte count and longer telomeres identifies a subgroup who show excellent overall survival (OS) after IST.8 The presence of a paroxysmal nocturnal hemoglobinuria (PNH) clone is associated with better response in some but not all studies.
Specific treatment of refractory AA
Unrelated donor HSCT
Unrelated donor HSC transplantation (UD HSCT) is indicated after failure of one course of IST for patients who are fit enough for HSCT and who have a suitably matched UD.9-11 For children, the benefit of UD HSCT over a second course of IST was reported from Japan. The 5-year OS was similar between the 2 groups, but failure-free survival was significantly better (84%) for those receiving transplantations compared with those who received further IST (9%) and most had continuing pancytopenia.12 UD HSCT for SAA is discussed in depth by Professor Socié elsewhere in this publication.13
Treatment options if there is no suitable UD
The difficult situation encountered is when a patient with refractory SAA has no suitably matched UD, and this is not uncommon, especially in those of minor ethnic origin or mixed ethnicity. The options for these individuals include a second course of IST, an alternative immunosuppressive drug or novel agent, or an experimental form of transplantation using an alternative donor source, namely cord blood or a haploidentical family donor. There is no consensus on the optimal approach and the treatments may be challenging. Issues to consider at this stage when deciding whether to advise a nontransplantation or transplantation approach include patient age, comorbidities, and patient and family wishes. Although IST carries a lower short-term treatment-related mortality than transplantation, the major long-term concern is clonal evolution, the risk of which is increased with repeated courses of ATG. Conversely, although HSCT offers the chance of cure, the main risks of alternative donor HSCT remain graft rejection and GVHD, especially chronic GVHD, which affects mortality and quality of life. We have summarized our choice of possible options in the Figure 1 algorithm.

Algorithm for the management of refractory AA patients: our personal perspective.
Algorithm for the management of refractory AA patients: our personal perspective.
Repeat course of ATG (Table 1).
The clinical experience reported frequently is the use of rabbit ATG to salvage patients failing to respond to an initial course of horse ATG. Although the second course of IST with rabbit ATG + CSA is associated with response rate between 22% and 77% in different series,14,15 response is inferior compared with the responses (50%-60%) seen in the relapsed setting,15 possibly indicating extreme HSC exhaustion in refractory AA. The addition of a third immunosuppressive agent or growth factor to further augment the response rates seen with ATG + CSA as first-line therapy has been disappointing.16-18 A retrospective study from National Institutes of Health (NIH) assessed response to rabbit ATG + CSA in 22 patients refractory to initial horse ATG + CSA–based regimens. The median age was 31 years and the median interval between the 2 ATG courses was 205 days.15 A response rate of 30% was seen and 1000-day survival was 70%, with median OS not reached. Clonal evolution was evident in 18% (2 MDS and 2 acute myeloid leukemia). A randomized controlled trial from the same institution compared rabbit ATG + CSA (n = 27) versus alemtuzumab (n = 27) in refractory AA, and found comparable response rate (33% vs 37%, P = .78).19 In the rabbit ATG arm, the 3-year cumulative incidence of relapse and clonal evolution was 19% and 16%, respectively. OS at 3 years was 60% and was not statistically different from the alemtuzumab group. An Italian multicenter study14 evaluated rabbit ATG + CSA as a second-line therapy after failed horse ATG + CSA in 30 AA cases. G-CSF at dose of 5 μg/kg subcutaneously from days 1 to 90 was also administered. Median age was 21 years and the median interval between the 2 ATG courses was 151 days. Transfusion independence was achieved in 77% patients with 30% complete response and OS was 93% with a median follow-up of 914 days. The higher response rate seen in this study compared with the NIH study could be related to the shorter time between 2 cycles of ATG, implying a delayed response to first course, and also to the different response criteria used. A third course of ATG is invariably ineffective in refractory AA.7
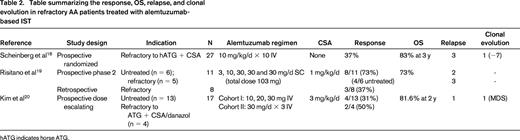
Other immunosuppressive drugs: alemtuzumab (Table 2) and cyclophosphamide.
Alemtuzumab, a humanized anti-CD52 IgG1 monoclonal antibody, is used in a wide range of conditions, including autoimmune cytopenias, allogeneic HSCT conditioning, AA, and pure RBC aplasia.10,19 A pilot study20 by the European Group for Blood and Marrow Transplantation (EBMT) analyzed 19 AA (including 13 refractory) patients treated with alemtuzumab administered subcutaneously (total dose 103 mg) and low-dose CSA (1 mg/kg) with a response rate of 58%. Infectious complications were not a major issue and this was possibly due to the strict administration of antimicrobial prophylaxis. Treatment was administered in the outpatient setting. Although initial responses were encouraging, early relapses were frequent (7 of 9), with some patients responding to rechallenge with a single dose of alemtuzumab. Late relapses (22%) and clonal evolution (15%) were not an infrequent event. The NIH study investigated the role of alemtuzumab monotherapy (10 mg IV for 10 days) in refractory, relapsed, and treatment-naive AA.21 For refractory disease, patients were randomized between rabbit ATG + CSA (n = 27) versus single-agent alemtuzumab (n = 27) and the 6-month response rates (37% for alemtuzumab) were comparable between the 2 arms; 3-year OS was 83%. The regimen was well tolerated and infectious complications were minimal. Avoidance of CSA in this protocol makes it a feasible option for older patients, especially those with renal impairment or CSA intolerance. The heterogeneity of response, 56% in the relapsed setting, 37% in refractory disease, and 19% in treatment-naive AA, is of interest from the pathobiology point of view. In the same study, 10 additional patients who were refractory to both horse and rabbit ATG received alemtuzumab and response was seen in 2 patients. A dose-escalating study21 of alemtuzumab (60 and 90 mg) in combination with CSA in 19 AA (4 refractory) patients induced a response of 35%; unexpectedly, all responses occurred in the 60 mg arm. The ease of administration, its efficacy even without concurrent use of CSA, and the relatively good safety profile makes alemtuzumab a good alternate choice of IST in relapsed/refractory AA for salvaging transplantation-ineligible patients.
High-dose (50 mg/kg × 4 days) cyclophosphamide (CY) has been used in both upfront and salvage therapy for AA, and a pilot single-institution study suggested comparable responses to horse ATG/CSA with fewer relapses and less clonal evolution. Long-term follow-up data (10 years)22 showed that the OS rate was 62%, the response rate was 48%, and the actuarial event-free survival rate 27% in 23 refractory patients. Although the early mortality rate was 7.5%, the cumulative incidence of fungal infections was very high at 40% in refractory patients in view of prolonged cytopenia (median time to response 5 months). An NIH prospective study comparing high-dose CY with “standard arm” (ATG + CSA) in treatment-naive patients, was terminated prematurely in view of excess deaths and increased incidence of fungal infections in the CY arm.23 Moderate-dose (120 mg/kg) CY for untreated AA induced meaningful responses but showed significant toxicity in a study presented at the ASH annual meeting in 2012.24 In view of its significant toxicity, the use of CY should be restricted25 to clinical trials because less-toxic drugs are currently available.
Alternative donor transplantation options: cord blood transplantation and haploidentical HSCT.
The largest studyof unrelated cord blood transplantation (CBT) in acquired SAA is reported by the combined EUROCORD/EBMT Severe Aplastic Anemia Working Party (SAAWP), comprising 71 patients with SAA (9 with PNH) and median age of 13 years (range 2-68). 26 A reduced-intensity conditioning (RIC), fludarabine-based regimen was used in 68% patients; 57 received a single unit and 14 a double unit CBT. The main problem was engraftment failure, with a cumulative incidence of neutrophil recovery of only 51% at 2 months and a 3-year OS of 38%. Better engraftment (58%) and OS (45%) was observed in recipients of > 3.9 × 107 total nucleated cells (TNCs)/kg. All those patients receiving total body irradiation (TBI) 12 Gy as part of the conditioning regimen died, indicating that a RIC rather than myeloablative regimen is preferable. Similar results were reported from the Japanese Registry12 ; in that study, of 31 patients with median age of 28 years (range, 0.9-72), the cumulative incidence of neutrophil recovery and platelet recovery was 55% and 72%, respectively. The 2-year OS was 41%, although a small subgroup receiving low-dose TBI (2-5 Gy) with fludarabine and CY had the best OS of 80%. A smaller study from Japan evaluated a RIC regimen of fludarabine, melphalan, and TBI 4 Gy in 12 adults (median age 49 years; range, 20-70).27 Median TNC at cryopreservation was 2.5 × 107/kg (range, 1.8-4.4). There was 1 primary graft failure in a patient with HLA antibody against the donor cells and 1 late graft failure at 3 years. Ten patients are still alive.
The optimal conditioning regimen for CBT in SAA is not known. A RIC regimen should be used incorporating fludarabine. Because of the high risk of nonengraftment, a higher cell dose (EBMT recommendation is to use > 4 × 107 TNCs/kg) is required compared with doses used in CBT for leukemia and with no more than a 2/6 HLA mismatches in the cord unit(s). In addition, patients should be screened for HLA antibodies that may be directed against HLA antigens present on the cord units and thereby increase the risk of rejection, so that only cord units lacking that antigen(s) are avoided.28 The current EBMT protocol comprises fludarabine 30 mg/m2 × 4, CY 30 mg/kg × 4, ATG, and TBI 2 Gy, with CSA alone as postgraft immunosuppression.29 Because CBT is still experimental, it should only be performed as part of prospective clinical study.
A recent novel approach is to coinfuse cord blood with haploidentical CD34+ cells to aid engraftment after CBT. In a small (n = 8) study presented at the ASH annual meeting in 2012, early T-cell engraftment was of cord blood origin; myeloid engraftment was initially from the haploidentical stem cells, followed by dual engraftment of both cord blood and haploidentical cells and then loss of haploidentical donor cells with full donor myeloid engraftment from the cord cells.30 Larger studies are indicated to further assess this approach.
The key features of haploidentical related HSCT are that a graft is available for most patients, the time to procure the graft is short, and the cost is low. However, the presence of HLA antibodies in the recipient that are directed against a haploidentical family member will likely preclude the use of that donor. This is not a new approach to HSCT for SAA patients lacking a matched sibling donor or a suitably matched UD. Historically, haploidentical HSCT was invariably unsuccessful, with high rates of graft rejection and GVHD. A recent review by the EBMT SAAWP on 73 patients receiving transplantations between 1976 and 2011 and mostly using nonmyeloablative regimens showed a 3-year OS of only 37%.31 Disappointingly, there was no improvement in OS with procedures performed even after 1999, highlighting the need for a new approach to conditioning for haploidentical HSCT. A novel approach is nonmyeloablative conditioning with high-dose CY given on days +3 and +4 after transplantation to prevent GVHD by depleting dividing donor-alloreactive T cells but sparing quiescent, nonalloreactive T cells. High-dose CY is not toxic to the infused HSCs due to their high content of aldehyde dehydrogenase, which confers resistance to CY. Such an approach has been reported anecdotally in PNH and warrants further exploration in SAA.32 As for CBT, patients must be screened for HLA antibodies directed against the donor cells and, if present, an alternative haploidentical family donor must be chosen against whom no HLA antibodies are found.33 Attempts to desensitize the recipient pretransplantation may be successful, but not always.
Other agents: eltrombopag and androgens.
Eltrombopag an oral thrombopoietin mimetic licensed in chronic immune thrombocytopenic purpura that induces platelet maturation and release by binding to c-MPL receptors on megakaryocytes. In a recent phase 2 study of 25 patients with refractory SAA, eltrombopag induced a response (at 12 weeks) in at least one hematologic lineage in 44% patients and 36% no longer required platelet transfusions.34 Interestingly, 24% patients became RBC transfusion independent and 36% had a neutrophil response. Trilineage responses were seen in 24% of patients; although surprising, this might indicate stimulation of c-MPL receptors on remaining stem cells. The starting dose was 50 mg/d, with subsequent increases in 25 mg increments fortnightly in nonresponders to a reach a maximum of 150 mg. The drug was well tolerated and there were no reports of increased reticulin or collagen fibrosis in the BM, although clonal evolution with monosomy 7 was detected in 2 nonresponders. The safety and efficacy of eltrombopag needs to be evaluated further in prospective clinical trials, especially in view of the possible link to clonal evolution. The early and rapid response seen with eltrombopag has led to design of prospective trials with incorporation of this agent along with ATG + CSA for first-line treatment of AA.
Androgens such as oxymethalone have been used historically for the treatment of AA before the availability of ATG/CSA and subsequently as an adjunct to ATG35 and are still used in certain developing countries for first-line treatment of AA. Androgens lead to increased telomerase activity via aromatization of estradiol to steroids36 and thus induce responses in patients with telomeropathies who manifest as apparent acquired AA.
A recent prospective trial evaluated the efficacy of danazol, a synthetic anabolic steroid, in relapsed (n = 3) and refractory (n = 13) AA.37 All patients completed the 12-week low-dose regimen (300 mg/d) without side effects and with a response rate of 31%. Although side effects such as abnormal liver functions, virilization in females, and peliosis hepatitis were not reported, this could be due to the short treatment phase.
Supportive care in refractory SAA
What is the current best supportive care and why is it important?
The impact of best supportive care is relevant at all stages of the disease. The quality of supportive care at initial presentation of SAA is vital and will determine the survival when patients are frequently at high risk of bleeding and infection. Best supportive care continues through initial therapies, whether with HSCT or ATG. Because response to ATG is delayed until ∼ 3 months, best supportive care is critical during this time to help ensure optimal outcome.
Patients who respond to first-line IST have significantly better survival than nonresponders. However, the OS of refractory patients has also improved significantly over time (23% vs 35% vs 57% during time periods 1989-1996 vs 1996-2002 vs 2002-2008, respectively)38 ; there was a significant reduction in deaths from infection including invasive fungal infections, emphasizing the importance of improvements in supportive care in refractory patients (Figure 2).

Improvement in survival of refractory AA patients over time. The data indicate the outcomes for 174 patients with SAA who were unresponsive to initial IST at 6 months. Three patient groups were identified, group 1 (n = 43); December 1989-October 1996; group 2 (n = 51, November 1996-October 2002); and group 3 (n = 80, November 2002-April 2008).The first column indicates survival curves censored for HSCT and the second graph is not censored for HSCT. A significantly improved 5-year OS for nonresponders to IST was seen in group 3 compared with other groups. Adapted and modified with permission from Valdez et al.38
Improvement in survival of refractory AA patients over time. The data indicate the outcomes for 174 patients with SAA who were unresponsive to initial IST at 6 months. Three patient groups were identified, group 1 (n = 43); December 1989-October 1996; group 2 (n = 51, November 1996-October 2002); and group 3 (n = 80, November 2002-April 2008).The first column indicates survival curves censored for HSCT and the second graph is not censored for HSCT. A significantly improved 5-year OS for nonresponders to IST was seen in group 3 compared with other groups. Adapted and modified with permission from Valdez et al.38
Transfusion issues
Alloimmunization after transfusion
RBC and platelet transfusions should be administered to maintain safe blood counts, but they should not be given unnecessarily. Most centers administer prophylactic platelets at a count of < 10 × 109/L or < 20 × 109/L if fever or bleeding is present, rather than withholding platelets unless bleeding occurs. RBC and platelet transfusions should not be avoided for fear of either alloimmunization or iron overload in the case of RBC transfusions.
Despite leukodepletion of blood products, HLA alloimmunization in AA remains a problem and may occur through indirect or direct allorecognition. The consequences of alloimmunization are platelet transfusion refractoriness most often due to HLA antibodies (less often HPA antibodies) and graft rejection after HLA-identical HSCT due to minor HC antigens or HLA antibodies in the setting of mismatched HSCT. Platelet refractoriness is usually managed with HLA-matched platelets. AA patients with multispecific HLA antibodies are especially problematic because it may be difficult to obtain suitably matched platelet donors. In this situation, platelet units can be selected that are negative for the antigens recognized by the HLA antibodies; a new future approach may be the use of HLA epitope-matched platelets identified using the HLAMatchmaker program.39 Family members should be avoided as blood product donors for patients who are potential transplantation candidates, because this may cause sensitization to the donor minor HC antigens, resulting in increased risk of allograft rejection.
Should irradiated blood products be given to all AA patients?
Aside from the mandatory requirement for irradiated blood products for all patients undergoing HSCT, the rational for irradiated blood products in AA is to reduce the risk of transfusion-associated GVHD after ATG or alemtuzumab and possible allosensitization to HLA and non-HLA antigens, based on canine experiments. A survey from the EBMT reported that most centers give irradiated products after ATG but with no consensus on how long to continue. The EBMT SAAWP advised that it is reasonable to continue while the patient is still on CSA. Some centers have a universal policy for all AA patients regardless of type of treatment or if untreated or for all myeloid patients in general.40
When should iron chelation be started and what is the best drug to use?
There is no evidence base to establish when to start iron chelation therapy in multi-transfused AA patients and recommendations are extrapolated from other disorders such as MDS. Currently, ferritin of > 1000 μg/L is the proposed threshold to initiate chelation for potential transplantation candidates because iron overload contributes to transplantation-related mortality. For other patients, the need for iron chelation is made on an individual basis. The use of subcutaneous/IV desferrioxamine or oral deferasirox varies between centers and countries. Desferrioxamine is an effective iron chelator, although data in AA are lacking and compliance is often a big issue. Deferasirox is an oral iron chelator and is effective in AA, but may be dose limited by nephrotoxicity when used with CSA, and other side effects such as rash and gastrointestinal upset have occurred.41 Deferiprone is in general to be avoided due to a risk of agranulocytosis, but may be a useful alternative agent or may be used in combination with another iron chelator when there is an urgent need to reduce iron overload in refractory AA patients awaiting HSCT.
Infection is the major cause of death in SAA: what is best treatment and prophylaxis?
Infection is the major cause of death in SAA, in which neutropenia is often prolonged, especially in refractory patients, resulting in a high incidence of invasive fungal infection and systemic bacterial sepsis. Invasive fungal infection is a major cause of death, most often due to Aspergillus species; but the Zygomycetes (Mucor, Rhizopus), Candida, Fusarium, and other fungi with a predilection for the lungs, nasal sinuses, and brain may also be responsible. Stenotrophomonas maltophilia was the most frequent gram-negative infection in one study from MD Anderson Cancer Center; other important gram-negative pathogens in SAA include Pseudomonas, E coli, and Klebsiella species. Febrile neutropenia requires broad spectrum IV antibiotics, with early administration of systemic antifungal if fevers fail to respond promptly.
What is the current role of granulocyte transfusions?
We are now able to collect larger cell doses of granulocytes by apheresis and, with prior administration of a dose of G-CSF and dexamethasone, granulocyte transfusions may be potentially lifesaving in severe sepsis such as invasive fungal disease, particularly for patients due to proceed to HSCT. HLA alloimmunization was reported in 17% of patients42 ; patients who are transplantation candidates should be retested after granulocyte transfusions to ensure the absence of HLA antibodies directed against the potential donor.
What is the best prophylaxis for SAA?
Prophylaxis for SAA is a controversial issue and practice varies widely due to the lack of prospective studies. Many centers use prophylactic antibiotics and antifungals in SAA to help prevent gram-negative infections and invasive fungal infections, but other centers choose not to use antifungal prophylaxis and instead commence systemic antifungals early with febrile neutropenic episodes. Antiviral prophylaxis with acyclovir or valacyclovir should be used during and after ATG therapy. During ATG therapy, subclinical reactivation of CMV and EBV is common but self-limiting, and therefore does not require antiviral treatment. EBV posttransplantation lymphoproliferative disorder has only very rarely been reported after ATG, most often after rabbit ATG. It is not our practice to give Pneumocystis prophylaxis with ATG, but this is done routinely in some centers in the United States. Nebulized pentamidine is an appropriate drug for prophylaxis because cotrimoxazole is myelosuppressive.
Other aspects of management of infection in AA
There is no role for corticosteroids in the treatment of AA because they increase the risk of fungal and bacterial colonization. Their short-term use with ATG is only to help prevent serum sickness. The use of G-CSF for initial treatment of AA is not encouraged because it is not effective in treating AA and merely delays the onset of appropriate specific treatment, during which time the patient may have become infected and/or alloimmunized.
This article was selected by the Blood and Hematology 2013 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2013. This article is reprinted with permission from Blood. 2013; Volume 122.
Disclosures
Conflict-of-interest disclosure: A.G.K. declares no competing financial interests. J.C.W.M. has consulted for and received honoraria from Pfizer and Sanofi. Off-label drug use: Alemtuzumab, ATG, and oxymetholone as treatment for AA.
Correspondence
Judith C. W. Marsh, Department of Haematological Medicine, King's College Hospital, Denmark Hill, London SE5 9RS, United Kingdom; Phone: 44-203-299-3709; Fax: 44-203-299-3514; e-mail: judith.marsh@nhs.net.