Abstract
This review summarizes the available evidence and outlines our approach to the prophylaxis and management of posttransplantation lymphoproliferative disorder (PTLD) in adult solid organ transplantation recipients. We attempt to reduce immunosuppression as tolerated in every patient with suspected PTLD in close cooperation with their transplantation physician. There is no evidence to guide the decision when to initiate further treatment; we usually wait no longer than 4 weeks and always initiate further therapy unless there is a complete or at least good partial remission. If clinical and histological findings indicate rapidly progressive disease, we initiate additional therapy significantly earlier. CD20-positive PTLD accounts for approximately 75% of PTLD cases. Outside of clinical trials, we currently regard sequential therapy with rituximab and CHOP (cyclophosphamide, hydroxydaunorubicin, vincristine, prednisone/prednisolone) chemotherapy as standard evidence-based treatment for CD20-positive PTLD unresponsive to immunosuppression. We also discuss our approach to the rare instance of adults with PTLD associated with primary EBV infection, localized (stage I) disease, rare PTLD subtypes, and refractory/relapsed disease based on the available retrospective data and our own experience. In addition to immunotherapy and chemotherapy, this includes local therapy approaches such as surgery and radiotherapy in stage I disease, plasmacytoma-like PTLD, and primary CNS PTLD. We also provide our view on the current indications for the use of allogeneic cytotoxic T cells, even though this treatment modality is so far unavailable in our clinical practice.
Introduction
Posttransplant lymphoproliferative disorder (PTLD) is a group of lymphatic and plasmacytic proliferations arising in the setting of immunosuppression after transplantation. Histologically, their spectrum ranges from polyclonal early lesions to monomorphic lymphoma. EBV proteins or nucleic acids can often be demonstrated in tumor cells (Table 1), but cases occurring years after transplantation are frequently EBV negative. The aim of this review is to provide an account of our interpretation of the available evidence and our approach to the management of PTLD. Although we feel confident in answering the question “CD20-positive PTLD: what to do?” based on prospective trial data, this is not the case for rarer PTLD subtypes or refractory/relapsed disease. For such cases, we offer an account of what we (currently) do based on the available retrospective data and our experience with all of the limitations of an “expert opinion.” We hope this will aid physicians in making their own informed treatment decisions. Although referring to relevant evidence from PTLD in pediatric patients and PTLD after allogeneic hematopoietic stem cell transplantation, the scope of this review is limited to risk factors, prophylaxis, and management of PTLD after solid organ transplantation (SOT) in adult patients.
EBV: what to do?
Risk factors for PTLD after SOT
The incidence of non-Hodgkin lymphoma is significantly increased in patients after SOT and varies with the transplanted organ and the intensity of immunosuppression. It is highest after combined heart and lung transplantation and in patients receiving antilymphocyte antibodies for prophylaxis of graft rejection.1 Strikingly, in kidney transplantation recipients with graft failure and cessation of immunosuppression, lymphoma incidence reverts to pretransplantation levels.2 The incidence of PTLD after SOT is bimodally distributed, with early and late peaks; risk factors and histological findings, most notably the frequency of EBV association, differ between early PTLD (within the first year after transplantation) and late PTLD. This suggests separate mechanisms of lymphomagenesis.2,3 However, risk factors vary depending on the studied population. In a large cohort of Australian kidney transplantation recipients, significant risk factors for early PTLD included EBV seronegativity at transplantation and receipt of T-cell–depleting antibodies, whereas significant risk factors for late PTLD included time from transplantation, age, and use of calcineurin inhibitors.2 The evidence regarding the role of different classes of maintenance immunosuppressive agents in particular is inconclusive (for a more detailed review of the evidence, see Parker et al4 ).
PTLD prophylaxis after SOT
Strategies for PTLD prophylaxis in adults focus on the prevention and treatment of EBV reactivation. A large multicenter retrospective study of anticytomegalovirus immunoglobulin prophylaxis showed a reduced incidence of early PTLD in renal transplantation recipients.5 Another strategy is monitoring of peripheral blood EBV viral load, which has been linked to PTLD risk.6 A low incidence of PTLD could be demonstrated in pediatric liver and adult lung transplantation recipients whose immunosuppression was adjusted guided by EBV viral load.7,8 In adult heart transplantation recipients, the combination of EBV viral load monitoring and preemptive rituximab treatment has also shown a reduced PTLD incidence compared with historical controls.9
PTLD: EBV-specific therapy approaches
Antiviral therapy in PTLD associated with primary EBV infection.
There is insufficient evidence of efficacy and safety to recommend the use of antiviral agents outside of clinical trials.10,11 In our clinical practice, there is one exception: EBV-seronegative SOT recipients (common in children, rare in adults) are at very high risk of PTLD associated with primary EBV infection: 20%-30% in adults receiving an organ from an EBV-positive donor (donor positive, recipient negative).12 These patients have neither humoral nor cellular immunity to EBV infection. In this instance, we have had positive experiences with antiviral therapy. We have previously described successful treatment of PTLD after primary EBV infection with cidofovir and IV immunoglobulins in a young liver transplantation recipient with PTLD refractory to rituximab and CHOP (cyclophosphamide, hydroxydaunorubicin, vincristine, prednisone/prednisolone) chemotherapy.13
EBV-specific cytotoxic T cells.
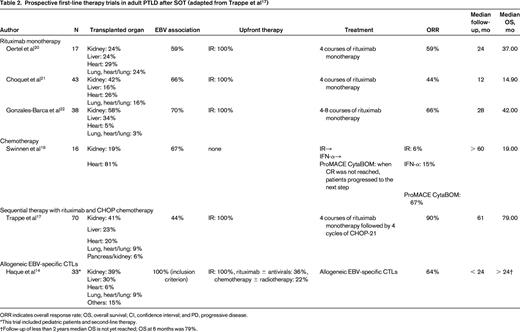
The use of ex vivo–derived EBV-specific cytotoxic T cells (CTLs) to treat EBV and EBV-associated PTLD has been successfully tested in a prospective clinical trial including patients after SOT (Table 2).14 In our current clinical practice, CTLs do not play a role due to their lack of availability in Germany. The main obstacle to this approach is the time needed for production of CTLs. Where readily available, we see a definite role for CTLs for the treatment of PTLD after SOT in primary CNS PTLD,15 refractory EBV-positive disease, and in patients with poor performance status. We currently do not support the use of CTLs as first-line treatment in CD20-positive PTLD due to the higher overall response rate of sequential immunochemotherapy in clinical trials and a lack of prospective overall survival data beyond 24 months (Table 2). Evidence for and future strategies in CTL technology have recently been reviewed in depth.16
PTLD with or without EBV: what to do?
Treatment of PTLD after SOT
PTLD is a rare disease and encompasses a wide spectrum of both histological (Table 1) and clinical presentations. Clinically, treating PTLD is challenging due to its heterogeneity and a limited evidence base. Longstanding immunosuppression results in a high risk of infectious complications, concomitant diseases often include limited renal or cardiac function, and extranodal and/or disseminated manifestations are common. Prospective phase 2 trial data have been published in the largest subgroup, CD20-positive B-cell PTLD, accounting for roughly 75% of cases. For rarer histological entities and primary CNS PTLD, data are only available from case reports and retrospective case series.
Staging investigations
Our routine work-up for patients with PTLD includes history and examination with a focus on performance status and comorbidities, imaging (computed tomography of neck, thorax, abdomen and pelvis; in cases of limited renal function, magnetic resonance imaging), as well as laboratory investigations assessing hepatic and renal function, differential blood count, and serum lactate dehydrogenase activity. We strive to have all biopsy specimens reviewed by a hematopathologist with experience in the field of PTLD. Work-up should always include BM biopsy to assess infiltration by PTLD (approximately 10% of cases).17 We perform bowel ultrasonography with the goal of detecting gastrointestinal manifestations and, in case of clinical suspicion or positive Hemoccult test, we also request gastroscopy and colonoscopy. Virological investigations include EBV, HIV, and hepatitis serology, as well as EBV viral load; we evaluate hepatitis antigens and viral load in case of serological suspicion or previously documented infection. Serology results from the transplantation center should be requested and reviewed. We do not routinely perform positron emission tomography.
Immunosuppression reduction
Immunosuppression reduction (IR) has the goal of reestablishing host T-cell function sufficiently to control lymphoproliferation without compromising the grafted organ and is applicable to all subtypes of PTLD. Established 30 years ago, high response rates (45%) have been reported in a retrospective analysis.18 Without doubt, PTLD including monomorphic and EBV-negative cases can respond to IR. The response rates, however, have not been reproduced prospectively so far, with the only prospective trial conducted demonstrating a response to IR in 1 of 16 cases (6%).19 We attempt to reduce immunosuppression as tolerated in every patient with suspected PTLD in close cooperation with their transplantation physician. Detailed guidance is available in the comprehensive British interdisciplinary guidelines.11 There is no evidence to guide the decision as to when to initiate further treatment; we usually do not wait for longer than 4 weeks and initiate further therapy unless there is a complete or at least good partial remission. If clinical and histological findings indicate rapidly progressive disease, we initiate additional therapy without delay (Figure 1).

Local therapy approaches (surgery/radiotherapy)
Radiotherapy and surgery have a role as additional treatment modalities synchronous or in sequence with systemic therapy. In addition, radiotherapy and surgery combined with immunosuppression reduction can offer a curative approach for PTLD localized to a single site (Ann Arbor stage I). A retrospective analysis of 30 patients treated with surgery and IR reported a 3-year overall survival of 65%.18 Nevertheless, and despite a lack of supporting evidence, we favor adjuvant rituximab treatment in case of CD20-positive disease. The central role of local therapy approaches in plasmacytoma-like PTLD and primary CNS PTLD will be discussed below.
First-line treatment of PTLD
CD20-positive PTLD
Rituximab.
The efficacy and safety of the anti-CD20 antibody rituximab as first-line treatment has been tested in CD20-positive PTLD unresponsive to immunosuppression reduction in 3 prospective clinical trials (Table 2). Two independently performed, multicenter, prospective phase 2 trials administered rituximab monotherapy at a dose of 375 mg/m2 weekly for 4 consecutive weeks. Complete remission (CR) rates were 9/17 (52%) and 12/43 (28%), respectively.20,21 However, a combined analysis of both trials demonstrated that 34/60 patients (57%) experienced disease progression within 12 months of completing rituximab therapy, including 9/35 (26%) of treatment responders. Interestingly, this was the case in half the patients with a partial response (5/10), but in less than a fifth of patients with a CR (4/25). Thirty-two of those 34 patients received further treatment including chemotherapy, further rituximab monotherapy, and radiotherapy. Median overall survival was 34.5 months, with 1- and 2-year overall survival rates of 72.5% and 51.8%, respectively. A prospective, multicenter, phase 2 trial from Spain tested extended treatment with rituximab.22 Patients who did not achieve CR after 4 weekly infusions of rituximab received a second course of 4 rituximab infusions. The CR rate of 13/38 (34%) after the first course improved to 23/38 (61%) with a second course of rituximab and overall survival was 47% at 27.5 months. In summary, single-agent rituximab as first-line treatment after immunosuppression reduction is effective and safe with virtually no clinically relevant infectious toxicity; however, CR rates are approximately 35% and early disease progression is common. Extended therapy with rituximab may increase the CR rate, but whether this translates into improved overall survival is unclear (Table 2).
Chemotherapy.
First-line chemotherapy without rituximab in PTLD has to our knowledge only been tested in a single prospective phase 2 trial (Table 2). Of 7 patients unresponsive to reduction of immunosuppression and IFN-α, 5 (71%) had a complete response to chemotherapy with ProMACE CytaBOM (cyclophosphamide 650 mg/m2 day 1, doxorubicin 25 mg/m2 day 1, etoposide 120 mg/m2 day 1, prednisone 60 mg/m2 days 1-14, cytosine arabinoside 300 mg/m2 day 8, bleomycin 5 mg/m2 day 8, vincristine 1.4 mg/m2 day 8, and methotrexate 120 mg/m2 day 8 repeated every 21 days for a total of 6; supportive care was filgrastim 5 μg/kg/d on days 2-14 or until recovery of WBC); and 1/7 (14%) suffered treatment-related mortality (TRM) due to an infectious complication.19 Four of the 5 complete responders were disease-free at their 2-year follow-up.
The key finding, namely that chemotherapy can induce lasting remissions in PTLD at the price of toxicity, including TRM, in approximately 20% of patients is supported by retrospective analyses. For example, Elstrom et al reported an overall response rate of 74% (57% CR) in 23 patients receiving chemotherapy.23 The majority of patients received CHOP (n = 10) or R-CHOP (rituximab + CHOP; n = 9), median overall survival was 42 months, and TRM 6/23 (26%). Fohrer et al reported an overall response rate of 24/33 (73%) in PTLD treated with dose-adjusted ACVBP (doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone), 60% 5-year overall survival, and a TRM of 3/33 (9%).24 Nine of 33 patients had received rituximab either before or in parallel with chemotherapy. A retrospective analysis from our own study group showed an overall response rate of 17/26 (65%), a median overall survival of 14 months, and 31% TRM in 26 patients receiving first-line CHOP chemotherapy.25
Sequential therapy.
In 2003, the European PTLD Network initiated the PTLD-1 trial, an international phase 2 trial combining first-line rituximab and CHOP in fixed sequence (NCT01458548). The goal was to improve the long-term efficacy of rituximab therapy and to avoid the toxicity of CHOP seen in first-line treatment. In summary, SOT recipients with CD20-positive PTLD unresponsive to reduction of immunosuppression received 4 weekly courses of 375 mg/m2 rituximab followed by 4 weeks without treatment and 4 cycles of CHOP-21 chemotherapy (cyclophosphamide 750 mg/m2 IV day 1, doxorubicin 50 mg/m2 IV day 1, vincristine 1.4 mg/m2 IV day 1, and prednisone 50 mg/m2 orally on days 1-5) at 3-week intervals starting at day 50. In case of disease progression under rituximab treatment, patients proceeded to chemotherapy immediately (therefore starting before day 50). Supportive treatment included mandatory G-CSF support and antibiotic prophylaxis (cotrimoxazole and ciprofloxacin were recommended). Key exclusion criteria were CNS involvement, HIV infection, severe organ dysfunction not related to PTLD, and Eastern Cooperative Oncology Group (ECOG) performance status > 2. Of 70 patients assigned to sequential treatment, 76% had late, 96% monomorphic, and 44% EBV-associated PTLD. Main adverse events were grade 3/4 leukopenia in 68% and grade 3/4 infections in 41% of patients. Seven (10.6%) of the 66 patients receiving CHOP suffered TRM. Five of 7 CHOP-related deaths occurred in rituximab nonresponders and included Pneumocystis jirovecii pneumonia before obligatory prophylaxis had been introduced, biliary hemorrhage, and 3 cases of bacterial sepsis. Two rituximab responders died from pulmonary hemorrhage and hepatitis C reactivation. The overall response rate was 90%, with 67% complete responses; 74% of responders were progression free at 3 and 5 years. With 6.6 years median overall survival, the PTLD-1 trial, the largest prospective phase 2 trial in the field to date, has demonstrated the efficacy and safety of sequential therapy (rituximab followed by CHOP chemotherapy).17 Key points are the improved overall response rate from 60% at interim staging after rituximab to 90% after sequential therapy (20% and 68% complete responses, respectively) and a longer median overall survival compared with the European rituximab monotherapy trials (1.2, 3.1, and 3.5 years; Table 2).20-22 In addition, we found sequential therapy to be equally effective in EBV-positive and EBV-negative PTLD despite on average poorer performance status and more common chemotherapy dose reductions in patients with EBV-positive PTLD. TRM in rituximab responders was lower (2/41, 4.9%) than in nonresponders (5/25, 20%), suggesting lower chemotherapy toxicity in patients with lower tumor burden. Furthermore, a prospective trial of a protocol combining rituximab and low-dose chemotherapy in pediatric PTLD also had favorable results.26 Therefore, we currently view sequential therapy as standard evidence-based treatment for CD20-positive PTLD unresponsive to immunosuppression reduction outside of clinical trials.
Burkitt PTLD.
Burkitt PTLD is a rare form of CD20-positive monomorphic B-cell PTLD, accounting for less than 5% of PTLD cases overall. Like Burkitt lymphoma, it is characterized by extranodal manifestations, rapid growth, a very high proliferative index, and the presence of an MYC translocation in most but not all cases.27 Treatment of Burkitt lymphoma outside of the posttransplantation setting involves high doses of alkylating agents, frequent dosing, and intrathecal and/or systemic CNS prophylaxis.28 In adult Burkitt PTLD, however, CRs have been reported with treatment ranging from immunosuppression reduction plus rituximab monotherapy to immunosuppression reduction plus high-dose chemotherapy protocols. The latter approach is associated with significant mortality in adults (3/5, 60%).29 In a case series from the German PTLD study group, 5/8 patients received immunosuppression reduction followed by sequential immunochemotherapy (4 courses of rituximab followed by 4 cycles of CHOP or R-CHOP) and 5/5 patients reached a CR. There was no TRM and only 1 of these 5 patients suffered a relapse that could be successfully treated with rituximab plus carboplatin/etoposide (R-CE; see relapsed/refractory disease below).30 Based on this experience, we currently treat Burkitt PTLD with sequential therapy; however, due to the clinically aggressive course, we do not wait for the effect of immunosuppression reduction before initiating rituximab therapy. The role of intrathecal prophylaxis in the rituximab era is unclear.
Primary CNS PTLD.
Involvement of the CNS occurs in approximately 10% of PTLD cases, most commonly as primary CNS PTLD.31 A retrospective analysis of 84 cases of primary CNS PTLD from Australia, Europe, and the United States identified an association with renal transplantation (66/84). Most cases occurred as late PTLD (70/84 later than 1 year; 25/84 later than 10 years), had CD20-positive diffuse large B-cell lymphoma histology (66/84), and were EBV positive (74/79).32 The observation of late, EBV-associated PTLD differs markedly from systemic PTLD. Treatment was highly variable and included immunosuppression reduction, high-dose methotrexate, high-dose cytarabine, rituximab, whole-brain radiotherapy, and combinations. Although patients who achieved a response to therapy had a significantly improved prognosis, the optimal treatment regime is still unclear. It was interesting that in the above-mentioned analysis, patients who received immunosuppression reduction as sole initial treatment did particularly poorly. Furthermore, receiving rituximab and/or high-dose cytarabine was associated with a trend toward better overall survival in univariate analysis, but not in multivariate analysis. To put the result in context, it should be noted that the power of the multivariate analyses was limited by the diverse combinations of therapy and that high-dose cytarabine therapy was usually given subsequently to IV methotrexate. Overall survival in patients treated with immunotherapy, chemotherapy, and radiotherapy was approximately 30% after 3 years. In light of these poor results and the common EBV association of primary CNS PTLD, early treatment with CTLs (where available) is a reasonable approach. In patients with adequate renal (transplantation) function, we presently use immunosuppression reduction with concurrent rituximab and high-dose IV methotrexate (up to 4 g/m2) until a CR is reached. Nephrotoxicity affecting the graft (in conjunction with reduced immunosuppression) can result in allograft loss.33 For those patients with impaired renal function and/or ECOG > 2, we administer immunosuppression reduction, concurrent rituximab, and whole-brain radiotherapy. To reduce the incidence of infections, we furthermore advocate reducing high-dose steroids started to treat edema associated with intracranial space-occupying lesions as soon as feasible after initiation of definitive treatment, and to administer P jirovecii prophylaxis.
CD20-negative PTLD
Plasmacytoma-like PTLD.
Plasmacytoma-like PTLD is another rare subtype of PTLD accounting for approximately 4% of cases. Based on the highly congruent findings of the 2 largest adult case series (8 and 9 patients, respectively), key features include CD138 positivity and CD20 negativity, light chain restriction, presence of a serum paraprotein, and EBV association in approximately half the cases. In contrast to plasma cell neoplasms in the immunocompetent host, BM involvement and lytic bone lesions are rare, whereas extranodal, often isolated plasmacytomas are the norm.34,35 From a treatment perspective, long-lasting CR can be achieved in localized disease with the combination of immunosuppression reduction and radiotherapy. In cases of disseminated disease, we have had positive experiences with immunosuppression reduction followed by combination chemotherapy containing bortezomib and doxorubicin (PAD), in analogy to multiple myeloma therapy in the immunocompetent patient.34 Overall outcome is favorable.
Hodgkin and Hodgkin-like PTLD.
Hodgkin and Hodgkin-like PTLD form a separate group in the World Health Organization classification of PTLD.36 Published data on treatment outcomes is scarce and limited to case reports and small retrospective series.37,38 The picture is further complicated by the histological subtypes of Hodgkin lymphoma and the different management of limited versus disseminated disease in immunocompetent patients. Consistent with published guidelines11 and mirroring treatment of Hodgkin disease in the immunocompetent host, we use immunosuppression reduction followed by radiotherapy in stage I disease and systemic chemotherapy (ABVD [doxorubicin, bleomycin, vinblastine, dacarbazine]) as tolerated in stage II, III, or IV disease. It should be noted that ABVD in PTLD is associated with significant (infectious) mortality both in our own experience and in the published case series.37,38
Plasmablastic PTLD.
Plasmablastic lymphoma (PBL) was first described as a usually EBV-associated B-cell neoplasm with immunoblastic morphology, the immunophenotype of plasma cells and loss of mature B-cell antigens arising in the oral mucosa of HIV-positive patients. It is clinically characterized by extranodal, particularly mucosal, manifestations; a highly aggressive course; and poor prognosis. In the largest case series to date (8 cases) of PBL PTLD, we observed an association with heart or heart/lung transplantation. Furthermore, median time to PTLD was 12.8 years, the longest observed in any PTLD subtype so far. Immunosuppression reduction and local therapy were not sufficient to treat PBL PTLD even in localized disease, whereas the combination of immunosuppression reduction and systemic chemotherapy (CHOP-21) could achieve lasting CRs in 3/8 patients (2/3 with localized disease, 1/5 with disseminated disease). However, successful treatment was only possible in lymphomas that were both EBV associated and negative for the MYC/IGH translocation.39 Our current treatment approach for PBL PTLD is therefore immunosuppression reduction and early CHOP-21 chemotherapy with growth factor support and P jirovecii prophylaxis.
T-cell PTLD.
The final subgroup of rare PTLD cases to be discussed here is T-cell PTLD. PTLD of T-cell origin is associated with poor prognosis. The largest case series published so far consists of 9 patients,40 and “meta-analyses” of published cases are available.40,41 Although T-cell PTLD cases are now relatively well characterized as a histologically heterogeneous group and clinically as occurring late (median time to PTLD, > 60 months), with extranodal manifestations in most patients and EBV association in approximately one-third, little is known about the best treatment approach. Swerdlow identified T-large granular cell leukemia as a prognostically favorable subgroup,41 which we can confirm from our experience. In the series by Herreman et al, the only patient surviving beyond 2 years was a 13-year-old kidney transplantation recipient with ALK-negative anaplastic large cell lymphoma treated with combination chemotherapy.40 One patient with refractory hepatosplenic T-cell lymphoma underwent allogeneic stem cell transplantation, which achieved a CR of PTLD; however, the patient succumbed to fungal infection within months. Due to the paucity of evidence, our current management of T-cell PTLD is based on immunosuppression reduction and combination chemotherapy, most commonly CHOP based.
Treatment of relapsed/refractory non-CNS PTLD
For PTLD refractory or relapsed after immunosuppression reduction, rituximab in case of CD20-positive disease, and cytotoxic chemotherapy, no prospective trial data are available. In our experience, these patients often have a poor performance status and are at even higher risk of infection under chemotherapy than those in the first-line situation. Published retrospective data are also scarce. Our study group has reported a response to rituximab in 4/7 relapsed adults pretreated with chemotherapy with or without rituximab.42 In an analysis conducted in the pre-rituximab era, 9 patients either refractory (7 patients) or relapsed (2 patients) after first-line chemotherapy were treated with CE (carboplatin AUC4 d1, etoposide 120 mg/m2 on days 1-3 for 21 days). Five of 9 achieved a CR, whereas 2/9 died due to treatment complications.43 However, we doubt that response rates will be this high in patients pretreated with sequential chemoimmunotherapy. In our current clinical practice, we use R-CE in refractory PTLD or early relapses (within the first year) if the patient is fit for chemotherapy. We view CTLs as an important alternative in this situation where available. In case of later relapses (after the first year), we favor retreating in analogy to the first-line situation. Finally, and despite case reports of treatment successes, in our experience, the toxicity of autologous or allogeneic stem cell transplantation outweighs its benefit.
Common complications of PTLD (treatment)
Because treatment complications, particularly infections, are a significant source of morbidity and mortality in PTLD treatment, prophylaxis has a key role. Due to the risk of hepatitis B reactivation, all patients with previous hepatitis B and PTLD should receive prophylaxis consisting of lamivudine and IV immunoglobulins.44 During chemotherapy, we use G-CSF support as primary prophylaxis of febrile neutropenia. In the PTLD-1 trial, prophylaxis of P jirovecii pneumonia was recommended after 3 cases had occurred within the first year of recruitment (960 mg cotrimoxazole orally 3 times a week),17 and we now administer cotrimoxazole to all PTLD patients undergoing chemotherapy. We also use ciprofloxacin prophylaxis from chemotherapy until the patients has recovered from neutropenia. In the PTLD-1 trial alone, 3 cases of TRM were due to bacterial sepsis. An additional problem is posed by the gastrointestinal disease manifestations common in PTLD: gastrointestinal perforation, often due to excellent response to treatment.45 Sadly, the outcome of fulminant peritonitis in a PTLD patient in pancytopenia after chemotherapy is disastrous. Even though we have no evidence to support this measure, we consider resection of residual intestinal PTLD manifestations before starting chemotherapy. After successful therapy with chemoimmunotherapy, B-cell depletion and hypogammaglobulinemia often persist for at least 1 year. In patients with hypogammaglobulinemia and recurrent infections after successful PTLD treatment and in those undergoing second-line therapy, we administer IV immunoglobulin infusions as infection prophylaxis. Patients with initially poor performance status (ECOG > 2) are usually excluded from clinical trials. Those who were inadvertently included into the PTLD-1 trial despite an ECOG of 3 had a very high rate of treatment complications.
Future directions
For rare subtypes of PTLD and refractory PTLD, there is still a dire need for more clinical studies, even if they are “only” series of uniformly treated patients, regardless of the outcome. For CD20-positive B-cell PTLD, there are many open questions. Response to rituximab monotherapy at interim staging was predictive of both time to progression and overall survival in the PTLD-1 trial.17 So can patients who achieve a CR with rituximab monotherapy be spared chemotherapy without risking a higher relapse rate or poorer overall survival? Is there a benefit to R-CHOP chemotherapy? We are currently testing these hypotheses in a prospective trial (risk-stratified sequential treatment, RSST; NCT00590447). Interim results have been promising.46 Additional future stratifiers might include the differential blood count,47 prognostic scores (unpublished data), or molecular markers.
Summary: PTLD, a treatment algorithm
In Figure 1, we have outlined the structure of our current approach to the management of adult PTLD after SOT. We want to emphasize that PTLD requires close interdisciplinary cooperation involving the transplantation physician, pathologist, hematologists, radiation oncologist, surgeon, primary care physician, and, if applicable, the palliative care physician. Furthermore, the patient numbers required to produce meaningful trial or registry results can only be achieved by close international cooperation.
All of our patients with PTLD after SOT receive immunosuppression reduction guided by the transplantation physician. This should be initiated once there is high clinical suspicion of PTLD. Cases of PTLD after primary EBV infection are unique in that we use a trial of antiviral therapy. The delay between immunosuppression reduction and initiation of further therapy depends on histological subtype and the pace of disease progression. We wait longest in early lesions, stage I polymorphic and diffuse large B-cell lymphoma PTLD, and plasmacytoma-like PTLD; conversely, we start additional treatment in Burkitt PTLD, primary CNS PTLD, PBL PTLD, and T-cell PTLD as soon as a definitive histological diagnosis is available. The main distinction in management is CD20-positivity: CD20-positive cases are eligible for rituximab treatment. However, our approach varies with histology, stage, and clinical course, as detailed in the figure.
Disclosures
Conflict-of-interest disclosure: R.U.T. has received research funding from AMGEN, CSL Behring, Mundipharma, Roche, and Novartis; has consulted for Takeda and Roche; and has received honoraria from CSL Behring, Roche, and Novartis. H.Z. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Ralf Ulrich Trappe, German PTLD Study Group, Department of Hematology and Oncology, DIAKO Hospital Bremen, Gröpelinger Heerstrasse 406–408, 28239 Bremen, Germany; Phone: +49-421-6102-1481; Fax: +49-421-6102-1439; e-mail: rtrappe@gwdg.de; http://www.lymphome.de/Gruppen/DPTLDSG/index.jsp.