Abstract
In recent years, research in molecular genetics has been instrumental in deciphering the molecular heterogeneity of acute myeloid leukemia (AML), in particular the subset of patients with “intermediate-risk” cytogenetics. However, at present, only the markers NPM1, CEBPA, and FLT3 have entered clinical practice. Treatment of intermediate-risk AML patients eligible for intensive therapy has not changed substantially. The “3 + 7” induction therapy still represents the standard of care. The addition of the immunoconjugate gemtuzumab ozogamicin to therapy has been shown to improve outcome; however, the drug is not approved for this use. A common standard for postremission therapy is the administration of repeated cycles of intermediate- to high-dose cytarabine. Allogeneic stem cell transplantation may offer a survival benefit for many patients with intermediate-risk AML. Patients are best selected based on the genetic profile of the leukemia cells and the risk associated with the transplantation itself. A myriad of novel agents targeting mutant leukemia drivers or deregulated pathways are in clinical development. In the past, many novel compounds have not met expectations; nonetheless, with the rapid developments in comprehensive molecular profiling and new drug design, there is the prospect of personalizing therapy and improving patient outcome.
Learning Objectives
To understand that AML with “intermediate-risk” cytogenetics is a very heterogenous group of patients who now can be divided based on a large number of gene mutations
To understand that, at present, only the molecular markers NPM1, CEBPA, and FLT3 have entered clinical practice
To understand that, to achieve progress, all patients should be entered in clinical trials and, for correlative studies, BM and blood samples should be stored in a biobank
To understand that novel therapies are in clinical development that target specific mutant driver proteins and deregulated pathways
Introduction
Cytogenetic analysis to identify chromosome abnormalities has become well established in the clinical management of patients with acute myeloid leukemia (AML).1 These chromosomal abnormalities have been used to categorize patients into 3 risk groups: favorable, intermediate, and adverse.2-5 With the development of the novel genomics technologies, numerous new mutations or gene expression signatures have been identified that now allow us to decipher the molecular heterogeneity of AML, in particular within the large subset of “intermediate-risk” AML.6 With the advent of these technologies, it has become evident that AML is characterized by a remarkable genetic heterogeneity, with individual patients presenting with a distinct and almost unique combination of chromosome changes and somatically acquired gene mutations.
Despite these tremendous advances in our understanding of the disease pathogenesis, translation of these insights into the clinical setting lags behind and our current paradigm of therapy for AML has not changed substantially. In daily practice, the decision algorithm follows 2 major assessments: (1) whether a patient is eligible for intensive standard anthracycline and cytarabine (“3 + 7”)-based induction therapy and (2) which type of postremission therapy should be applied. Among younger adult intermediate-risk patients, the second point mainly relates to the question of whether a patient should be assigned to allogeneic hematopoietic stem cell transplantation (HSCT).
One factor that should be kept in mind is that our current genetic risk strata have been primarily developed in younger adult patients (18 to 60 or 65 years of age). Although this risk categorization in general has also some validity in older patients, outcome of these patients has remained very poor (Figure 1) and differences among genetically defined subsets of the disease become rather marginal. Therefore, this review mainly focuses on the impact of the novel molecular markers on current and future treatment concepts in younger intermediate-risk patients.

OS for intermediate-risk AML by age group. Shown in black are patients ≤60 years of age (n = 1188); in red are patients >60 years of age (n = 493). These data were obtained from 1681 intermediate-risk AML patients treated within the AMLSG treatment trials AMLHD93, AMLHD98A (www.ClinicalTrials.gov identifier NCT00146120), AMLHD98B, AMLSG 06-04 (www.ClinicalTrials.gov identifier NCT00151255), and AMLSG 07-04 (www.ClinicalTrials.gov identifier NCT00151242).
OS for intermediate-risk AML by age group. Shown in black are patients ≤60 years of age (n = 1188); in red are patients >60 years of age (n = 493). These data were obtained from 1681 intermediate-risk AML patients treated within the AMLSG treatment trials AMLHD93, AMLHD98A (www.ClinicalTrials.gov identifier NCT00146120), AMLHD98B, AMLSG 06-04 (www.ClinicalTrials.gov identifier NCT00151255), and AMLSG 07-04 (www.ClinicalTrials.gov identifier NCT00151242).
Intermediate-risk AML definition based on conventional cytogenetics and molecular genetics
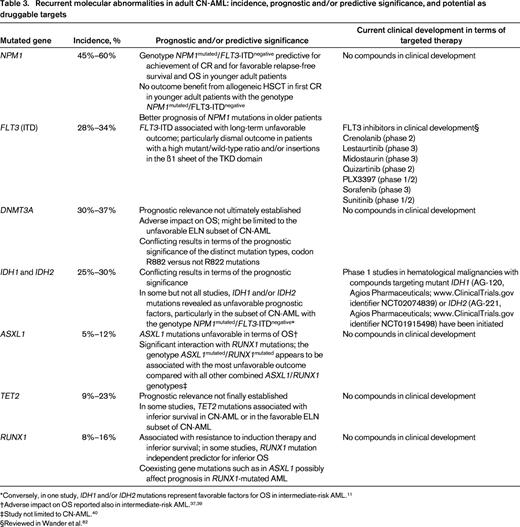
Based on chromosome abnormalities, patients with AML traditionally have been categorized into 3 risk groups: favorable, intermediate, and adverse (Tables 1, 2).1-5 The majority of AML patients have an intermediate cytogenetic risk, with most of them exhibiting a normal karyotype (Figure 2). In the recommendations by the European LeukemiaNet (ELN), for the first time, the 3 molecular markers NPM1 (nucleophosmin), CEBPA (CCAAT/enhancer binding protein (C/EBP), alpha), and FLT3 (fms-related tyrosine kinase 3) internal tandem duplications (ITDs) have been incorporated into this scheme, thus dividing the large group of cytogenetically normal (CN)-AML into molecular favorable and unfavorable subsets (Table 1). In terms of CEBPA mutations, there are emerging data showing that only double, not single, CEBPA mutations confer a favorable prognosis.7-10 Within AML with single mutated CEBPA, other concurrent molecular markers such as NPM1 and/or FLT3-ITD mutations affect outcome and, depending on the present NPM1/FLT3-ITD genotype, this subset of CEBPA-mutated AML has a more or less favorable long-term outcome.10 Over the last years, novel genetic data have been accumulating rapidly. Many of the molecular markers, such as DNMT3A (DNA (cytosine-5-)-methyltransferase 3 alpha), RUNX1 (runt-related transcription factor 1), ASXL1 (additional sex combs like transcriptional regulator 1), IDH1 (isocitrate dehydrogenase 1 (NADP+), soluble), IDH2 (isocitrate dehydrogenase 2 (NADP+), mitochondrial), and TET2 (tet methylcytosine dioxygenase 2), are most prevalent in the intermediate-risk cytogenetic group.11 For some of the mutations, data with respect to prognosis are quite consistent, whereas for others, the picture is less clear (Table 3). At present, none of these novel markers is practice changing. In the following, selected markers that allow dissection of intermediate-risk AML are discussed briefly; the impact of these markers has been best studied in CN-AML (Table 3).

Pie chart illustrating the distribution of the most frequent cytogenetics defining intermediate-risk AML according to the ELN recommendations.1 These data are based on 1681 intermediate-risk AML patients treated within the AMLSG AMLHD93, AMLHD98A (www.ClinicalTrials.gov identifier NCT00146120), AMLHD98B, AMLSG 06-04, and AMLSG 07-04 (www.ClinicalTrials.gov identifier NCT00151242) studies.
Pie chart illustrating the distribution of the most frequent cytogenetics defining intermediate-risk AML according to the ELN recommendations.1 These data are based on 1681 intermediate-risk AML patients treated within the AMLSG AMLHD93, AMLHD98A (www.ClinicalTrials.gov identifier NCT00146120), AMLHD98B, AMLSG 06-04, and AMLSG 07-04 (www.ClinicalTrials.gov identifier NCT00151242) studies.
NPM1 mutation.
Mutations in NPM1 have emerged as one of the most important molecular markers in AML. With an overall incidence of 25%–35% (CN-AML, 45%–60%), NPM1 mutations represent the most frequent gene mutation in AML.12-14 NPM1 mutations have been associated with chemosensitivity in younger and older patients.14-17 In younger patients, inferior prognosis is conferred by the concurrent presence of FLT3-ITD, whereas the impact of other co-occurring lesions, such as DNMT3A and IDH1/IDH2 mutations, is less clear. The availability of genetic testing for minimal residual disease has become another clinically relevant tool with which to identify patients with NPM1-mutated AML who are at high risk for relapse.18,19
FLT3-ITD mutation.
FLT3-ITDs are found in ∼20% of all AML (CN-AML: 28%–34%) and have been associated with inferior outcome.14,20 With respect to prognosis, there are 2 important issues: the mutant-to-wild type ratio and the ITD insertion site.21-23 Data on the allelic ratio consistently show an association between high allelic burden,20,21 which implicates in general a threshold of >0.50, and unfavorable outcome.24,25 The prognostic impact of low and intermediate FLT3-ITD allelic ratio is discussed controversially.20,21,24,25 Beyond the allelic ratio, ITD insertion, particularly in the B1 sheet of the tyrosine kinase domain (TKD) 1 that is present in ∼1/4 of the cases, has been shown to be associated with very poor prognosis.22,23 Whether a concomitant NPM1 mutation adds to prognostication in AML with FLT3-ITD is currently a matter of debate. Gale et al reported a better outcome in FLT3-ITD-positive patients harboring a concurrent NPM1 mutation,24 whereas others showed that the “protective effect” of NPM1 in AML with higher FLT3-ITD allelic ratio (≥0.50) is diminished or completely lost.25
DNMT3A mutation.
DNMT3A mutations are detected in 18%–22%26-28 of all AML cases and in 30%–37% of CN-AML cases.26-29 In CN-AML, first studies reported on unfavorable impact of these mutations on overall survival (OS).26,27 In one further study limited to CN-AML, DNMT3A mutations were associated with inferior disease-free survival and in trend also with a shorter OS.29 Subset analyses in this study suggested an age-dependent impact of different DNMT3A mutation types.29 DNMT3A mutations not affecting codon R882 were associated with worse outcome in younger patients, whereas mutations at codon R882 had adverse prognostic impact in older patients.29 In our study, we did not see a prognostic impact of DNMT3A mutations in younger CN-AML as a whole group.28 However, we could show that these mutations conferred a significantly shorter OS in the subset of younger CN-AML patients classified as unfavorable according to the ELN recommendations.1 Another aspect of our study was the opposite clinical effect of the different mutation types in terms of relapse-free survival and OS; mutations at codon R882 were unfavorable in terms of relapse-free survival and mutations not affecting codon R882 were favorable in terms of OS. The results of another study in younger AML patients have suggested that the unfavorable effect of DNMT3A mutations could be overcome by increasing the dose of daunorubicin during induction therapy.11
IDH1 and IDH2 mutation.
Approximately 15%–20% of all AML cases and 25%–30% of CN-AML cases carry either IDH1 or IDH2 mutations.30 IDH mutations in AML cluster to distinct codons, namely IDH1 codon R132 and IDH2 codons R140 or R172.30 Several studies assessing the prognostic relevance of IDH1 and IDH2 mutations in CN-AML have yielded conflicting results. In some,31-33 but not other,34,35 studies, IDH1 and/or IDH2 mutations were revealed as an unfavorable prognostic factor in the subset of CN-AML cases with the genotype NPM1mutant/FLT3-ITDnegative. Conversely, one study in AML with intermediate-risk cytogenetics found IDH1 and IDH2 mutations to be a favorable factor for outcome, resulting in a 3-year OS above 80% in this genotype.11 Further subset analyses in this study revealed that the favorable effect of IDH2 mutations was found exclusively in patients with IDH2 R140 mutations; IDH2 R172-mutated alleles only rarely co-occured with NPM1 mutations.11 Therefore, the improved prognosis in IDH2/NPM1-mutated AML was mainly attributed to IDH2 mutations at codon R140,11 which is consistent with the findings from Green et al.36 Even less well established is the prognostic impact of IDH2 R172 in intermediate-risk and/or CN-AML. There is some evidence from a Cancer and Leukemia Group B (CALGB) study in CN-AML that these mutations might be associated with inferior induction success in this cytogenetic subset of AML patients.31
ASXL1 mutation.
ASXL1 mutations are found in 5%–11% of AML cases and in 5%–12% of CN-AML cases.37-40 The studies on ASXL1 mutations in AML are remarkably consistent with respect to the increasing incidence of these mutations with age (Figure 3)37-40 ; in CN-AML, ASXL1 mutations were detected at a 5-fold higher frequency in patients ≥60 years of age compared with those <60 years of age (16.2% vs 3.2%, respectively).38 In intermediate-risk AML and/or in the subset of CN-AML patients, ASXL1 mutations constitute an adverse prognostic factor for long-term outcome, with low complete remission (CR) rates found in all studies.37-40 Moreover, our recent study on ASXL1 mutations in AML shows a significant interaction of ASXL1 and RUNX1 mutations; the subset of patients with the genotype ASXL1mutated/RUNX1mutated had the lowest CR rate and the most unfavorable outcome compared with all other ASXL1/RUNX1 genotypes.40

Incidence of intermediate-risk AML associated gene mutations by age group. Age groups shown are: <45 years, 45–60 years, >60–75 years, and >75 years.
Incidence of intermediate-risk AML associated gene mutations by age group. Age groups shown are: <45 years, 45–60 years, >60–75 years, and >75 years.
TET2 mutation.
TET2 mutations are enriched in the subset of CN-AML patients, in whom their frequency ranges between 9% and 23%.41-43 The incidence of TET2 mutations in AML considerably increases with age (≤60 years: 7%–10%; >60 years: 19%–25%).44 Two studies have reported that TET2 mutations are unfavorable in terms of survival in CN-AML or in AML with intermediate-risk cytogenetics, but neither of these studies found TET2 mutations to be an independent prognostic factor after multivariable analyses.11,42 A CALGB study by Metzeler et al reported TET2 mutations as an adverse factor for CR achievement, event-free survival, and disease-free survival only among CN-AML defined as favorable risk according to the ELN recommendations.41 In contrast, in our study,43 we could not show a prognostic effect of TET2 mutations in either CN-AML patients or the ELN subsets. Thus far, there is no sufficient evidence for TET2 mutations as a clinically relevant prognostic marker in AML or in subsets of AML and a more comprehensive evaluation, in particular within the context of other potentially modulating genetic lesions,43 is necessary.
RUNX1 mutation.
Beyond involvement in recurrent chromosomal rearrangements, intragenic mutations of RUNX1 have been found in ∼5%-15% of AML cases.45-48 There is a significant increase of the mutation frequency with older age that is paralleled by an association of the mutation with secondary AML evolving from myelodysplastic syndrome (Figure 3). In all studies,45-48 RUNX1 mutations have consistently been associated with resistance to standard induction therapy and with inferior survival. In the studies by Tang et al45 and Mendler et al,48 RUNX1 mutations were revealed as a strong independent predictor for inferior OS. RUNX1 mutations are associated with a characteristic pattern of additional gene mutations. An intriguing finding is the frequent co-occurrence with mutations in genes encoding epigenetic modifiers such as ASXL1 (21%), MLL (alias KMT2A; lysine (K)-specific methyltransferase 2A) partial tandem duplication (17%), IDH2 (15%), and BCOR (BCL6 corepressor; 8%). The impact of such combined genotypes on prognosis is under investigation.40,49
Intermediate-risk AML therapy
Current standard
Induction therapy.
In patients eligible for intensive induction therapy, 3 days of an anthracycline (daunorubicin, 60 mg/m2, idarubicin, 10-12 mg/m2, or the anthracenedione mitoxantrone, 10-12 mg/m2) and 7 days of cytarabine (100-200 mg/m2 continuously IV) (3+7) still remains the standard of care in AML. With such regimens, CR is achieved in 60%–80% of younger adults and in ∼40%-60% of older patients. As a general theme, lower doses of anthracyclines (eg, daunorubicin 45 mg/m2 for 3 days) are no longer considered appropriate in either younger or older patients (at least up to the age of 65 years) based on data from 3 randomized trials comparing daunorubicin 45 mg/m2 and 90 mg/m2.50-52 Randomized trials comparing the current standard of daunorubicin (60 mg/m2) with high-dose daunorubicin (90 mg/m2) are ongoing.
In the studies by Fernandez et al50 [Eastern Cooperative Oncology Group (ECOG) E1900] and Lee et al52 of younger adult patients, explorative subset analyses showed that the survival benefit of higher doses of daunorubicin was confined to the intermediate-risk group and was not observed in the favorable- and adverse-risk groups. In the ECOG E1900 trial, a significant interaction was found between DNMT3A mutational status and the daunorubicin dose, in that high-dose daunorubicin was associated with an improved survival among patients with DNMT3A mutations.11 In the study by Löwenberg et al51 of older patients, exploratory post hoc analyses showed that patients 60–65 years of age had the greatest benefit from an escalated dose of daunorubicin; however, an interaction between treatment and intermediate-risk cytogenetics was not found.
For induction therapy, no other intervention has been shown convincingly to be better, with one potential exception: the addition of the immunoconjugate gemtuzumab ozogamacin (GO) to standard induction. In 3 randomized trials, the addition of single or fractionated doses of GO during induction (and consolidation) improved outcome of younger53 and older patients.54,55 In all 3 trials, it appears that patients with favorable- and intermediate-risk cytogenetics, including those with CN-AML, had a significant benefit from GO.53-56 To date, GO has not been approved again for the treatment of AML and is currently only available within clinical trials. However, the accumulated data on the efficacy of GO in newly diagnosed AML, particularly in favorable- and intermediate-risk AML, may provide a basis for a reapproval.57
Consolidation therapy.
Postremission therapy in intermediate-risk patients comprises conventional consolidation and autologous and allogeneic HSCT.1,58 One current standard of conventional consolidation for younger adult patients, repeated cycles of high-dose cytarabine 3 g/m2 twice daily on days 1, 3, and 5, was established by a landmark study of the CALGB.59 Prospective, up-front randomized trials comparing combination postremission therapy with single-agent high-dose cytarabine in younger adult patients failed to show an improvement in survival end points.60-63 The optimal dose of cytarabine and the number of cycles have remained open issues. Early pharmacological studies suggest a saturation of arabinosylcytosine-5′triphosphate formation in leukemic blasts already at dosages of 200-250 mg/m2/h, corresponding to a single dose of 1 g/m2 IV infusion of 3 hours.64 Allogeneic HSCT is associated with the lowest rates of relapse; however, the benefit of allogeneic HSCT on overall survival may be offset by nonrelapse treatment-related mortality. Meta-analyses of clinical trials that prospectively assigned allogeneic HSCT versus alternative consolidation therapies for AML in CR1 on an intent-to-treat donor versus a no-donor basis showed that allogeneic HSCT offers significant OS benefit for patients with intermediate-risk AML.65-67 The value of allogeneic HSCT is being reassessed based on the novel molecular markers that profoundly affect prognosis within the intermediate-risk group. As an example, a study by the German-Austrian AML Study Group (AMLSG) showed that AML patients with the genotype NPM1mut/FLT3-ITDneg, who in the ELN classification are now considered to be the “favorable” subgroup, do not appear to benefit from allogeneic HSCT in CR114 ; conversely, there are markers, such as FLT3-ITD (Richard F. Schlenk, manuscript submitted May 2014),14 RUNX1 mutation,47 or EVI1 overexpression,68 for which a beneficial effect was shown in retrospective studies. With respect to FLT3-ITD, in several studies, allogeneic HSCT from either a matched-related or a matched-unrelated donor has been shown to result in an improved outcome compared with chemotherapy and autologous HSCT.14,69-71 However, most of these studies are comparisons with historical controls or as-treated analyses,25,71 whereas statistically more valid intent-to-treat approaches with donor versus no donor comparisons are rare.14,69 A recent analysis by AMLSG and a Spanish study indicate that the benefit of allogeneic HSCT performed in CR1 may be restricted to patients with an allelic ratio of ≥0.51 (Richard F. Schlenk, manuscript submitted May 2014).25 Allogeneic HSCT did not improve outcome of patients with a low allelic ratio, suggesting that, in these patients, the risk associated with allogeneic HSCT was not outweighed by its benefit. Therefore, an allogeneic HSCT in patients with a low allelic ratio may not be the primary recommended postremission therapy. For individual decision making, it is recommended to consider both the disease risk, as best assessed by the cytogenetic and molecular genetic profile, and the risk associated with the transplantation, as assessed by different scores that include comorbidity and other transplantation-related risk factors.72 In general, in younger patients with genetic intermediate-risk AML and low Hematopoietic Cell Transplantation-Specific Comorbidity Index (HCT-CI) or European Group for Blood and Bone Marrow Transplantation (EBMT) scores, allogeneic HSCT is considered the most appropriate postremission therapy. The situation becomes more complex in patients who have higher transplantation-related risk indices.
In patients >60 years of age, the value of intensive postremission therapy continues to be a debate. High-dose cytarabine (3 g/m2 per single dose) has been shown to be too toxic. In addition, there is no general consensus as to how many cycles of consolidation therapies should be given. With respect to molecular markers, among intermediate-risk patients, only NPM1 mutations have been shown to predict for a better outcome after intensive chemotherapy.17,73,74
Future options
Due to the genetic heterogeneity and complexity of AML, no novel drug is currently expected to show sufficient activity as a single agent that would justify significantly lowering the doses of our current induction and consolidation therapy in patients eligible for intensive therapy. For future treatment trials investigating novel agents, a reasonable backbone consists of 3+7-based induction and repetitive cycles of age-adapted doses of cytarabine in consolidation therapy. In the past years, a myriad of novel compounds have entered clinical development in AML. Some of these compounds have been developed expressly to target molecular lesions that are considered important driver mutations (Table 3). For such compounds, it is justified to a priori restrict eligibility to patients with the respective genetic lesion. For the majority of novel agents, however, we lack predictive markers. To identify predictive molecular response signatures, it should therefore be mandatory to store patients' BM and blood samples from diagnosis in a biobank that allows such retrospective marker studies.
NPM1.
Currently, there is no targeted molecular therapy available for NPM1-mutated AML, but there are efforts to target NPM1-mutation-associated altered transport mechanisms.75 Based on high CD33 expression levels, the anti-CD33 antibody GO appears to be an attractive therapeutic strategy. Although a subset analysis within the MRC AML15 trial showed no survival benefit for GO in NPM1-mutated AML,53 other trials showed a benefit for GO in the low- and intermediate-risk groups.76 Therefore, the impact of GO may need to be revisited in the light of concomitant mutations and single-nucleotide polymorphisms in CD33 that might affect treatment response.77 Similarly, based on 2 controversial reports the potential benefit of all-trans retinoic acid (ATRA) in NPM1mutated/FLT3-ITDnegative patients remains elusive, but a recent AMLSG study in younger AML patients again suggests a beneficial effect.78,79
FLT3 inhibitors.
Small-molecule FLT3 inhibitors have emerged as an attractive therapeutic option in patients with FLT3 mutations (Table 3). First-generation compounds such as midostaurin (PKC412), lestaurtinib (CEP-701), sunitinib (SU-11248), and sorafenib (BAY-43-9006), as well as second-generation agents such as quizartinib (AC220) and crenolanib, are already being tested in clinical trials.80,81 The clinical activity of early inhibitors was limited by a lack of selectivity, potency, and also unfavorable pharmacokinetic properties. When used as single agents, they have limited antileukemic activity, mostly showing only transient reduction of blood and BM blasts; first randomized trials evaluating these agents have been disappointing.82,83 In a randomized trial of 224 patients with FLT3-mutated AML in first relapse, lestaurtinib did not increase the response rate or prolong survival.83 In a randomized trial of 201 newly diagnosed older AML patients, the addition of sorafenib to induction and consolidation therapy did not improve outcome in all patients or in the small subset of 28 patients with FLT3-ITD; results of induction therapy were worse in the sorafenib arm, with higher treatment-related mortality and lower CR rates.82 Results from the NCRI AML 17 trial (ISRCTN55675535) evaluating lestaurtinib and from the RATIFY trial (www.ClinicalTrials.gov identifier NCT00651261) evaluating midostaurin in newly diagnosed younger adult patients with activating FLT3 mutations are eagerly awaited.
The second-generation compounds such as quizartinib have improved potency and selectivity. This is associated with higher rates of BM blast clearance, however, mostly without achieving formal CRs. In addition, response duration is limited by the development of secondary resistance.84 A phase 2 trial in relapsed/refractory AML has been completed (www.ClinicalTrials.gov identifier NCT00989261) and a pivotal phase 3 study has been started comparing quizartinib monotherapy with salvage chemotherapy in relapsed/refractory patients with FLT3-ITD-positive AML (www.ClinicalTrials.gov identifier NCT02039726). Crenolanib is another highly selective and potent FLT3 inhibitor exhibiting strong activity against FLT3-ITD and the FLT3/D835 point mutants. Phase 2 trials are ongoing (www.ClinicalTrials.gov identifier NCT01657682, www.ClinicalTrials.gov identifier NCT01522469) and a phase 3 trial in relapsed/refractory patients with FLT3 mutations is in the planning phase.
IDH inhibitors.
Recently, 2 compounds specifically targeting mutant IDH1 R132H (AGI-5198)85,86 and IDH2 R140Q (AGI-6780)87 were identified. In fully transformed cells with endogenous IDH1 mutations, the selective R132H-IDH1 inhibitor AGI-5198 showed a near-complete R-2HG inhibition.86 Ex vivo treatment of primary IDH2 R140 AML cells with the small molecule AGI-6780, which potently and selectively inhibits the tumor-associated mutant IDH2/R140Q, induced the differentiation of primary human acute myelogenous leukemia cells in vitro.87 Based on these promising data, 2 investigational drugs, AGI-120 (targeting mutated IDH1) and AGI-221 (targeting mutated IDH2) have recently entered phase 1 clinical trials (www.ClinicalTrials.gov identifier NCT02074839, www.ClinicalTrials.gov identifier NCT01915498) (Table 3). In both trials, patients with advanced hematologic malignancies who exhibited IDH1 (www.ClinicalTrials.gov identifier NCT02074839) or IDH2 (www.ClinicalTrials.gov identifier NCT01915498) mutations were included. First results from the phase 1 study with AG-221 were presented at the European Hematology Association (EHA) meeting in 2014.88 Eligible patients included relapsed or refractory AML, myelodysplastic syndrome, and elderly untreated AML. AG-221 was administered orally once or twice a day in continuous 28-day cycles. First data on efficacy of the drug were available in 10 patients, with 6 of them revealing an investigator-assessed objective response (CR, n = 3; CR with incomplete platelet recovery, n = 2; partial response, n = 1). The data on the efficacy of the compound did hold up in the update presented at EHA 2014. Consistent with preclinical models, marked differentiation of myeloblasts into mature forms was associated with response. These data provide early validation of mutant IDH2 as a therapeutic target in cancer.
MLL-rearranged AML and DOT1L inhibition.
Deregulation in several epigenetic regulators that modify DNA or histones have been implicated in MLL-fusion-driven leukemogenesis.89 One therapeutic target is the histone methyltransferase DOT1L, which has recently emerged as an important mediator of MLL-fusion-mediated leukemic transformation via the modification of histone H3 on lysine 79 (H3K79).90 A first specific small-molecule inhibitor of DOT1L showed promising antiproliferative activity that was remarkably selective for cell lines bearing MLL rearrangements.91 A first-in-human phase 1 trial testing the DOT1L inhibitor EPZ-5676 in advanced hematologic malignancies, including AML with MLL rearrangements, is ongoing (www.ClinicalTrials.gov identifier NCT01684150).
PLK1 inhibition.
Polo-like kinases (Plks) are a family of 5 highly conserved serine/threonine protein kinases that have been shown to play a key role in mitotic checkpoint regulation and cell division.92 Plk1 is overexpressed in a range of human cancers, including AML.93 Volasertib (BI 6727) is a low-molecular-weight, ATP-competitive kinase inhibitor that potently inhibits Plk1.94 A predecessor compound, BI 2536,95 was previously evaluated in older patients with relapsed/refractory AML and provided a first proof-of-principle of the potential therapeutic value of targeting Plk in patients with AML.96 In a recent randomized trial comparing low-dose cytarabine with or without volasertib in older patients ineligible for intensive therapy, the combination therapy doubled the response rate and even showed a signal for a survival benefit.97 A pivotal placebo-controlled phase 3 trial in this patient population is ongoing (www.ClinicalTrials.gov identifier NCT01721876) and a randomized phase 2 trial evaluating volasertib in combination with intensive therapy, including younger adult patients (18 years and older) with newly diagnosed AML, is in the planning phase (www.ClinicalTrials.gov identifier NCT02198482).
BET inhibition.
Further therapeutic targets of potential relevance in AML are represented by the bromodomain and extraterminal (BET) chromatin readers. Preclinical data in AML suggest efficacy of BET inhibition across several AML subtypes.98 An early phase 1 study in hematologic malignancies including AML with CPI-0610, a small-molecule inhibitor of BET, has recently been initiated (www.ClinicalTrials.gov identifier NCT02158858).
Perspectives
A new era of clinical research in AML has been entered. With the rapid development in novel genomics technologies, comprehensive molecular profiling will now become integral part of our clinical trials. Molecular profiling will move away from the analysis of single genes toward the study of gene panels, which will allow a more precise diagnosis and the identification of patient subsets with distinct gene signatures, particularly among patients with intermediate-risk cytogenetics. Such an integrative mutational analysis will be instrumental to identifying patients who will benefit from novel molecular-targeted therapies. Although it may sound trivial, more rapid progress will only be achieved if the physicians caring for patients with AML enter their patients in a clinical trial. To date, testing for NPM1, FLT3-ITD, and CEBPA mutations at diagnosis is recommended to further specify the intermediate-risk AML group as defined by cytogenetics and to guide treatment decisions. BM aspirates should been preferentially used for the molecular analyses; peripheral blood is only meaningful in patients with a sufficient proportion of circulating blasts. In NPM1-mutated AML, minimal residual disease monitoring is clinically relevant because NPM1 mutant transcript levels were shown to be significantly associated with clinical outcome and to allow the early identification of patients at a high risk of relapse. Applicability of NPM1 minimal residual disease monitoring for preemptive therapy is currently under investigation. Diagnostic testing for other molecular markers outside of clinical studies is not practice-changing yet and thus cannot be recommended. However, it is likely that the accumulating data on the novel molecular markers will affect upcoming revisions of risk classification systems. A better understanding of the role of these molecular lesions in AML biology will hopefully result in the development of novel therapeutic approaches and improvement of clinical outcome for our AML patients.
Acknowledgments
This work was supported in part by Grant BU 1339/5-1 from the Deutsche Forschungsgemeinschaft and Grant 109675 by the Deutsche Krebshilfe. We thank Daniela Späth for support with creating the figures in this manuscript and Hartmut Döhner for critically reviewing the manuscript.
Disclosures
Conflict-of-interest disclosures: K.D. has received research funding from Novartis. P.P. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Konstanze Döhner, MD, Department of Internal Medicine III, University Hospital of Ulm, Albert-Einstein-Allee 23, D-89081 Ulm, Germany; Phone: 49-731-500-45001; Fax: +49-731-500-45505; e-mail: konstanze.doehner@uniklinik-ulm.de.