Abstract
Thrombocytosis has a large number of potential underlying causes, but the dominant group of hematological conditions for consideration in this setting are the myeloproliferative neoplasms (MPNs). In this chapter, we consider several key linked questions relating to the management of thrombocytosis in MPNs and discuss several issues. First, we discuss the differential diagnosis of thrombocytosis, which myeloid disorders to consider, and practical approaches to the discrimination of each individual MPN from other causes. Second, there have been several major advances in our understanding of the molecular biology of these conditions and we discuss how these findings are likely to be practically applied in the future. Third, we consider whether there is evidence that thrombocytosis contributes to the complications known to be associated with MPN: thrombosis, hemorrhage and transformation to leukemia and myelofibrosis. Last, we review current ideas for risk stratification and management of essential thrombocythemia and polycythemia vera as the 2 entities within the MPN family that are most frequently associated with thrombocytosis.
Learning Objectives
To be able to review the causes of thrombocytosis
To be able to diagnose an MPN
To understand current advances in the field with reference to molecular abnormalities and challenges in therapy
Introduction
There is no doubt that an exponential expansion in our knowledge of the pathogenesis of myeloproliferative neoplasms (MPNs) has occurred in the past decade. This triggered a change in how these conditions are diagnosed and a growing breadth of different therapeutic options. This chapter discusses management issues in MPNs starting with diagnosis and our current treatment algorithms for these conditions. We focus in particular upon essential thrombocythemia (ET) and polycythemia vera (PV) rather than on myelofibrosis (MF), because the latter is less commonly associated with thrombocytosis. Thrombocytosis is often the first clue to the diagnosis of MPN, but whether it truly contributes to disease-related complications and thus merits being treated is not fully clear. Treatment strategies for MPN vary from watchful waiting, aspirin, venesection, a range of cytoreductive therapies to allogeneic stem cell transplantation or the choice of an expanding number of ongoing clinical trials. For PV and ET, some very fundamental questions are outstanding, including the events surrounding transformation to MF and myeloid leukemia, better stratification for thrombosis, and whether hydroxycarbamide or hydroxyurea (HU) or IFN-α is the “best” first-line cytoreductive therapy.
Diagnosing an MPN
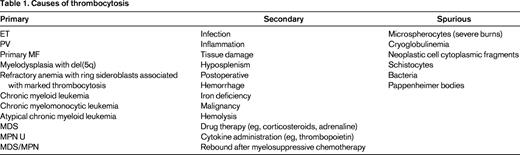
Naturally the most important initial step for management is to achieve an accurate diagnosis; an algorithm used in our practice is shown in Figure 1. For MPN, exclusion of reactive conditions has been simplified with the availability of reasonably specific molecular abnormalities, as exemplified by the recent description of Calreticulin mutations (CALR).1,2 The range of reactive or secondary causes specific to thrombocytosis is wide and is reflected in Table 1, where these are divided into primary, secondary, and spurious. Overall reactive causes are more common, although this depends upon the particular setting and patient features. In children, for example, a reactive cause is more likely than in the adult patient and persistence of the abnormality is also particularly useful. Having excluded a reactive disorder, it is ideal to use as much diagnostic information as possible to be able to assign the patient to a particular category of MPN. Although the MPN-unclassified category exists, there is no available information to facilitate management recommendations for this entity. Achieving an accurate MPN diagnosis relies upon the careful integration of data from several sources, including clinical evaluation, basic blood parameters (in particular the blood film(, molecular markers, cytogenetic analysis, and histology. The inclusion of cytogenetic or FISH analysis is particularly important when chronic myeloid leukemia and myelodysplasia need to be excluded and remains of prognostic significance for MF and possibly also PV.3 The entity of prefibrotic fibrosis remains controversial; further international collaboration and educational efforts are publically acknowledged to be required in this area.4 When prefibrotic MF is definitively present, it should likely best be managed as MF and seems to be associated with a worse prognosis. Whether prognostic scores, developed for use in MF, are appropriate for patients with a diagnosis of pre-fibrotic MF, or that after ET or PV, remains unclear.

Algorithm for diagnosis of MPN. CML indicates chronic myeloid leukemia; LDH, lactate dehydrogenase; and MDS, myelodysplasia. *Clear evidence of erthrocytosis refers to either hematocrit >0.56 in women or 0.60 in men, elevated RBC mass, or, in the absence of a secondary cause, low erythropoietin, JAK2 exon 12- or 14-positive, and hematocrit >2.5 SDs outside of the normal range.
Algorithm for diagnosis of MPN. CML indicates chronic myeloid leukemia; LDH, lactate dehydrogenase; and MDS, myelodysplasia. *Clear evidence of erthrocytosis refers to either hematocrit >0.56 in women or 0.60 in men, elevated RBC mass, or, in the absence of a secondary cause, low erythropoietin, JAK2 exon 12- or 14-positive, and hematocrit >2.5 SDs outside of the normal range.
Advances in molecular biology
Molecular abnormalities are a central feature in the World Health Organization (WHO) diagnostic criteria for MPN. With the discovery of the CALR mutations, a substantial proportion of patients will have a mutation in this gene, exon 12 or 14 of JAK2, or exon 10 of MPL. These mutations are primarily used for diagnostic purposes, as reflected in Figure 1. Beyond this, a range of additional genes may require investigation in evaluating an erythrocytosis: for example, the erythropoietin receptor, hypoxia-inducible factor, proline dehydrogenases, and others.5 Familial disorders may also need to be considered and several additional mutations have been described in this setting (eg, JAK2V617I).6 Beyond diagnostics in MF, there may be a role for testing for other mutations in genes such as ASXL-1, EZH2, IDH1 and IDH2, and SRSF2, which were associated with worse survival and a greater likelihood of transformation to acute leukemia in a recent study, or limiting this testing to CALR with ASXL1.7,8 At present, however, screening for such mutations is neither routine practice nor incorporated into prognostic scores. For patients with ET, there appears to be an association between the presence of CALR mutations and a better prognosis and a lower risk of thrombosis.9 Information to date with regard to quantitation of mutant allele burden, particularly of JAK2V617F, suggests that, when difficulties of reproducibility and interassay variation are resolved, this may be very useful in the setting of monitoring for minimal residual disease after transplantation.10 However, whether serial monitoring of JAK2V617F allele burden is important in any other setting has yet to be prospectively tested and is currently being evaluated in clinical trials. In PV, a high (>50%) JAK2V617F allele burden has been reported to be associated with greater risk of developing post-PV MF.11
Is thrombocytosis harmful in MPN?
Understanding of the pathogenesis of thrombosis and hemorrhage in MPN has not kept pace with advances in the hemostasis field. Problems limiting research in this area are the prolonged follow-up required to generate quality clinical data, and numerous confounding factors such as the impact of comorbid conditions, imprecise cytoreductive therapy that does not just affect one cell lineage, and biological heterogeneity. Multiple factors are likely to contribute to the pathogenesis of thrombosis, including increased RBC mass (in PV), thrombocytosis, platelet and leukocyte activation, and the formation of platelet leukocyte aggregates, in addition to circulating prothrombotic and endothelial microparticles; this field was recently comprehensively reviewed.12
Evidence for the contribution of the platelet to thrombotic risk in MPN includes histological studies demonstrating platelet-rich arteriolar microthrombi with minimal fibrin, the exquisite sensitivity of symptoms such as erythromelalgia to aspirin, and that aspirin therapy in the ECLAP study was significantly associated with a lower risk of cardiovascular events (relative risk 0.72, 95% confidence interval = 0.53–0.9).13 Finally, the contribution of platelet count per se as a risk factor has been presumed from clinical observations that cytoreductive therapy reduces the incidence of thrombosis. However, cytoreductive therapy is not completely specific in just controlling the platelet count and also affects the leukocyte count or hematocrit and other factors. In an analysis from the PT-1 study, the relationship between vascular complications and 21 887 prospectively collected blood counts in patients with ET (diagnosed according to the PVSG criteria) was investigated. After correction for confounding variables, no association was seen between blood counts at diagnosis and future complications. However, having a platelet count outside of the normal range during follow-up was associated with a risk of major hemorrhage (P = .0005), but not thrombosis (P = .7). Conversely, an elevated leukocyte count during follow-up was correlated with both thrombosis (P = .05) and major hemorrhage (P = .01). These data imply that the aim of cytoreduction in ET should be to keep the platelet count, and arguably the leukocyte count, within the normal range.14 They also provide a potential explanation for the differential effect of HU and anagrelide in the high-risk arm of this trial, in which anagrelide-treated patients were more likely to reach a composite primary end point of major hemorrhage, thrombosis, or vascular death (P = .03) despite equivalent long-term control of platelet counts. Subgroup analysis showed that arterial thrombosis, major hemorrhage, and MF were each significantly increased in anagrelide-treated patients (P = .004, P = .008, and P = .01, respectively), but venous thrombosis was less frequent (P = .006).15 Interestingly, although hemorrhage has been presumed to be due to acquired von Willebrand disease and to correlate with the degree of thrombocytosis, the proof of this association has not been demonstrated. Data from the PT-1 study elegantly show a correlation between thrombocytosis and hemorrhage risk, but no analysis of VWF was performed. In general, we do not screen for acquired VWD in ET or PV to predict risk of hemorrhage or to treat it. It is important to note here that data from the ANAHYDRET study comparing HU and anagrelide in ET patients (defined using the WHO criteria) suggested no difference in rates of thrombosis for patients treated with the different drugs, and thus differ from the PT-1 study.16 This may reflect a difference in study power, patient populations (WHO ET vs PVSG ET), and the differential effect of aspirin, which was not used in the ANAHYDRET study.
Concerning transformation to events such as MF or leukemia after an original diagnosis of ET or PV, although there is a correlation between degree of thrombocytosis and BM reticulin grade,17 there are no convincing data to suggest that the degree of thrombocytosis predicts transformation to either MF or leukemia. However, differential effects of therapy on rates of transformation to MF, for example, as observed in the PT-1 study, suggest that manipulation of blood counts or other aspects of disease may be important in changing the risk of these late myeloid events; more research should be directed into this area.
Management of ET and PV
The management of ET and PV after diagnosis aims to reduce the risk of thrombosis, control disease-related symptoms, and, where possible, reduce the risk of progression; cure is not possible using current therapies.
Risk stratification
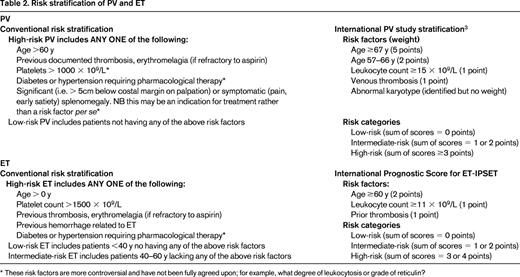
Beyond the generic management of vascular risks, patients should be assessed or risk stratified. Traditionally, this has been for thrombotic risk, although more prognostic scores address survival and leukemia risk. Several schemes have been proposed for this purpose (Table 2). Interestingly, quality-of-life issues are not featured in these scores, even though the burden of disease-related symptoms for ET and PV may be quite considerable and distinct patterns of such concerns are becoming more apparent.18 Evaluation of symptoms is an essential component of future studies and is becoming more important in the day-to-day clinical management of these patients. For patients with ET, the recently described International Prognostic Score (IPS ET)19 recategorizes a significant number of previously high- and low-risk patients. This is important because patients previously classified as low-risk have sustained major thrombosis and the converse is also true for high-risk patients. Unfortunately, IPS ET generates a large “intermediate-risk” category for which no treatment algorithm has yet been tested. Indeed, the “intermediate-risk” patient group in ET is controversial because it has been variably defined. Ongoing PT-1 trials will describe the natural history of ET and the response to aspirin versus aspirin plus HU for an intermediate-risk group defined by age 40-60 years with no high-risk features (ie, age <60 years, no prior thrombosis, platelets <1500 × 109/L) or cardiovascular risk factors. High-risk PV has in the past been less well defined than ET, but recent studies have improved this situation.3 Predominant factors are age, prior thrombosis, and leukocyte and platelet count (Table 2). Any novel markers for risk stratification should be robust and easily measurable. Current candidates include reticulin grade,17 JAK2V617F allele burden,20 and, for ET, the presence of the CALR mutation.2,9 The impact of conventional risk factors for atherosclerosis, especially if they are well controlled, is unclear.
Antiplatelet therapy
Low-dose aspirin is widely used for both ET and PV. However, although there is evidence from the ECLAP study for this strategy in PV,13 the use of low-dose aspirin remains controversial in ET. The ECLAP study enrolled 518 patients with PV who lacked a clear indication or contraindication for aspirin into a double-blind, placebo-controlled, randomized trial to assess the safety and efficacy of prophylaxis with low-dose aspirin (100 mg daily). The 2 primary end points for this study were: (1) the cumulative rate of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes and (2) these events plus pulmonary embolism and major venous thrombosis. After a follow-up of ∼3 years, the investigators reported that treatment with low-dose aspirin reduced the risk of the combined primary end point of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes compared with placebo, although the difference was not significant (P = .09). The use of low-dose aspirin also reduced the risk of the second combined end point to a statistically significant extent (P = .03). The incidence of major bleeding episodes was not significantly increased in the low-dose aspirin group (relative risk, 1.62; 95% confidence interval, 0.27-9.71). Interestingly, neither overall mortality nor cardiovascular mortality was significantly reduced; the trial was probably not adequately powered to address this question.13 In ET, the use of aspirin has never been addressed in a randomized controlled trial. Some studies, such as PT-1, included aspirin for all high-risk ET patients, whereas other more recent studies, such as ANAHYDRET, did not.15,16 This reflects a dichotomy of opinion in the field. These include concerns that bleeding is a particular risk for patients with very high platelet counts (see Is thrombocytosis harmful in MPN?) and perhaps that aspirin is not needed in patients whose disease is well controlled with cytoreductive therapy, although this has not been prospectively tested.
For “low-risk” ET, a retrospective analysis from a group of Spanish investigators suggests variable benefits for low-dose aspirin.21 In that study, the incidence rates of arterial and venous thrombosis were retrospectively analyzed in 300 low-risk patients with ET treated with antiplatelet drugs (mainly aspirin) as monotherapy (n = 198) or followed with careful observation (n = 102). Follow-up was 802 and 848 person-years for antiplatelet therapy and observation, respectively. Rates of thrombotic events were not different at 21.2 and 17.7 per 1000 person-years for antiplatelet therapy and observation, respectively (P = .6). Subgroup analyses suggested that JAK2V617F-positive patients not receiving antiplatelet medication showed an increased risk of venous thrombosis (P = .02) and that patients with cardiovascular risk factors had increased rates of arterial thrombosis while on observation (P = .047). Major bleeding was observed in patients with platelet counts >1000 × 109/L on antiplatelet therapy (P = .004). This retrospective analysis suggests that, for low-risk ET patients who are either JAK2V617F-positive or have cardiovascular risk factors, antiplatelet therapy has a benefit. In the remaining low-risk ET patients, aspirin does not appear to be effective for primary prophylaxis of thrombosis and observation may be an adequate option.
Phlebotomy
Concerning phlebotomy or venesection, the hematocrit target in PV has been controversial and was based upon a small series of patients in an elegantly conducted but old study.22 Data from the ECLAP study concluded that the hematocrit target of 0.45 was perhaps inappropriate.23 In a time-dependent multivariable analysis, a hematocrit in the evaluable range of 40%–55% was neither associated with the occurrence of thrombotic events or mortality nor with hematological progression. The investigators concluded that the range of hematocrit (<55%) encountered in the study population had no impact on the outcome of PV patients treated by current therapeutic strategies. This question has now been fully addressed in the recently reported CYTO PV study, in which PV patients were randomized to either a target hematocrit of 0.45 or 0.45-0.50. After a median follow-up of 31 months, the primary end point was recorded in 2.7% of patients in the low-hematocrit group and in 9.8% patients in the high-hematocrit group (P = .007). The primary end point plus superficial vein thrombosis occurred in 4.4% of patients in the low-hematocrit group, compared with 10.9% in the high-hematocrit group (P = .02). Therefore, the CYTO PV study strongly supports control of the hematocrit to at least 0.45, although, importantly, there were differences in leukocyte counts of the 2 arms of this study that may have accounted for some of the 44% reduction in thrombosis.24,25 There is no evidence to support a targeting the hematocrit <0.45, as has been suggested previously. A related but unanswered dilemma that arises in clinical practice is whether to control the hematocrit of a patient with a diagnosis of ET who has the JAK2V617F mutation and whose hematocrit is above 0.45, but ostensibly lies within the normal range. Masked PV should certainly be excluded in these patients. In practice, making this distinction would include use of RBC mass, but because this test is not widely available, other factors need to be considered. Such additional factors would include: careful review of BM histology, replenishment of iron stores, review of erythropoietin level, and testing for endogenous erythroid colonies; sometimes, careful observation until more obvious signs appear has to suffice.
Cytoreductive therapy
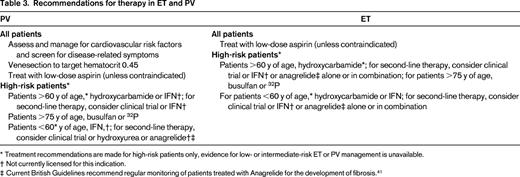
Regarding treatment targets, the European LeukemiaNet used a standard consensus approach to produce criteria for response; these criteria are designed primarily for clinical trials and also include reference to BM morphology and molecular markers.26 Appropriate first-line therapy for high-risk ET or PV is controversial; current published recommendations are summarized in Table 3. International practice varies widely, as demonstrated in the ongoing EXELS study with a recently published analysis of treatment patterns for high-risk ET across Europe,27 and there are many strong opinions regarding whether HU or IFN should be the treatment of choice. IFN and HU form a mainstay of therapy and there is an ongoing increase in interest in IFN, especially pegylated forms with disease-moderating effects. Pioneering studies with IFN were reported by Silver in 1988. Subsequent studies of pegylated IFN alpha-2a (Pegasys) in PV and ET report high levels of complete hematological response and good tolerance of the drug, particularly when started at low doses.28,29 There appears to be preferential targeting of the JAK2V617F-mutated clone by this agent.29 There are also data to suggest that Pegasys may be effective when other therapies have failed or have not been tolerated.30 A novel formulation of IFN from AOP pharma is also of interest in this context, with provisional results approximately equivalent to that for other formulations of IFN.31 Resolution as to which drug therapy is more appropriate for first-line therapy of ET and PV urgently requires evaluation in large randomized trials with comprehensive assessment of long-term side effects. Two such trials are currently ongoing internationally: MPD RC112, a phase 3 randomized study of HU versus Pegasys in newly diagnosed high-risk ET and PV, and PROUD-PV, a phase 3 study with AOP IFN in PV. It is absolutely critical that the field addresses the relative superiority of IFN and HU by completing the phase 3 trials because they will inform the relative benefits, efficacy, and toxicities of these medications.
The phenomenon of HU resistance or intolerance is important and identifies a group of ET and PV patients with a poor prognosis32 who require a change of treatment and for whom novel therapies may be attractive. Options for management in the face of HU resistance would include adjusting therapeutic targets (eg, to a platelet count of 600 × 109/L) or to switch to an alternative agent either alone or in combination. It must be remembered that HU, when used with or succeeded by agents such as busulfan, will significantly increase long-term risks of leukemia. For this reason, nonleukemogenic agents such as IFN or anagrelide are more appropriate in this setting. Patients with HU resistance or intolerance are the group of ET and PV patients for whom novel therapies are currently being evaluated.
Impact of JAK inhibitors and other novel therapies
There are data supporting the ability of some JAK inhibitors to control myeloproliferation in patients with PV and ET. However, beyond this, aspects that are uncertain include whether they prevent thrombosis and if they affect the probability of accelerated-phase disease such as MF or even leukemia. The size and duration of studies required to evaluate these aspects will be challenging. The first study in this context using CEP 701 (Cephalon) reported negative data; while there were responses in splenomegaly, pruritus, and phlebotomy requirements, CEP 701 treatment was not associated with a reduction in either leukocytosis or thrombosis and 5 thrombotic events occurred.33 In an initial report of the study using ruxolitinib (a JAK1 and 2 inhibitor) in 39 ET and 34 PV patients, there were reductions of splenomegaly and symptom scores; all patients had leukocyte counts <10 × 109/L, with 41% achieving platelet counts of <400 × 109/L, with no reported thrombotic events.34 This study was recently updated for PV patients,35 but no further data for the ET cohort is available. In the updated analysis, the PV cohort received ruxolitinib for a median of 152 weeks; a hematocrit <45% without phlebotomy was achieved in 97% of patients by week 24, with only one patient requiring a phlebotomy after week 4. Among patients with palpable splenomegaly at baseline, 44% and 63% achieved nonpalpable spleen measurements at weeks 24 and 144, respectively. Clinically meaningful improvements in pruritus, night sweats, and bone pain were observed within 4 weeks of the initiation of therapy and were maintained with continued treatment. Thrombocytopenia and anemia were the most common adverse events, thrombocytopenia of ≥grade 3 or anemia of ≥grade 3 occurred in 9% of patients each (1 patient had both) and were managed with dose modification.
Two large phase 3 commercially sponsored studies of ruxolitinib are fully recruited; the results of one (RESPONSE) were reported at the recent American Society of Clinical Oncology (ASCO) and European Haematology Association (EHA) meetings. Ruxolitinib was superior to best-available therapy for the composite primary end point of 35% spleen size reduction and freedom from venesection. This trial studied a highly selected group of patients who were intolerant or resistant to HU yet had splenomegaly and still required venesection. A confounding aspect is that >50% of the control arm was treated with HU, perhaps reflecting limited options for these patients. Most interesting was the reduction in thrombosis for ruxolitinib-treated patients. Further updates of this important data are awaited.36 There is one ongoing academic study of ruxolitinib in ET patients who are resistant or refractory to HU. Three important safety concerns have arisen with regard to JAK inhibition: a “withdrawal syndrome,” neurological toxicity, and risk of infections. An early report suggested a risk of severe inflammatory syndrome after ruxolitinib withdrawal and that these patients had a poor outcome.37 The COMFORT studies have not reported this as a risk, but a slow taper or consideration of steroid cover when ruxolitinib is withdrawn is suggested. A second JAK2 inhibitor, fedratinib, has recently been put on full clinical hold due to the development of complications similar to Wernicke's encephalopathy. Although this complication has not been reported with ruxolitinib, it will be important to evaluate other JAK inhibitors for this toxicity, especially because 2 other JAK inhibitors were also halted in development due to neurological toxicity.
Histone deacetylase inhibitors such as vorinostat38 and givinostat39 have been used in patients with ET and PV. Toxicities with these agents are variable and the administration of vorinostat in a phase 2 study led to a high discontinuation rate. Givinostat continues to be evaluated. The oral telomerase inhibitor imetelstat was investigated in ET, demonstrating molecular responses but also significant rates of hematological toxicity.40 Further studies of novel agents of sufficient duration are certainly needed to permit the evaluation of safety and efficacy.
Summary
It is very encouraging to witness recent developments in our understanding and treatment of MPN and to observe the benefits that these new options can provide to patients. Advances in our understanding of the molecular pathogenesis of these disorders combined with data from clinical trials underpin these improvements. There is an urgent need to complete studies to assess the relative benefits of HU and IFN as first-line therapies for both ET and PV. A greater understanding of the biological aspects of these diseases will hopefully lead to better defined surrogate markers for patients in need of novel therapies and to the development of more meaningful and practical trial end points.
Disclosures
Conflict-of-interest disclosures: C.N.H. has received research funding from Novartis; has consulted for Novartis, CTI, YM Bioscience, Gilead, and SBio; has received honoraria from Novartis and Sanofi; and has been affiliated with the speakers' bureau for Novartis, Sanofi, and Shire. N.C.G. declares no competing financial interests. Off-label drug use: JAK inhibition and other novel therapies such as Smo inhibitors for management of thrombocytosis.
Correspondence
Claire Harrison, Department of Haematology, Guy's and St Thomas' Foundation Trust, London SE1 9RT, UK; Phone: 442071882742; Fax: 442071882728; e-mail: Claire.Harrison@gstt.nhs.uk.