Abstract
Thrombotic microangiopathy (TMA) is a clinicopathological condition associated with a wide variety of medical conditions. TMA is classically characterized by microangiopathic hemolytic anemia, thrombocytopenia, and microvascular thrombi that cause end-organ damage. The most prominent diagnoses associated with TMA are thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Although TTP and HUS can have similar clinical and laboratory features and are often lumped together as a combined entity referred to as “TTP/HUS,” the pathologic processes causing TMA and optimal therapies for these conditions are different. Empiric use of therapeutic plasma exchange (TPE) in the setting of TMA is common. The high risk of morbidity and mortality associated with some causes of TMA justify rapid institution of this relatively low-risk procedure. However, many causes of TMA do not respond to TPE and prolonged courses of exchange in the absence of an underlying diagnosis may cause a detrimental delay in appropriate medical therapy. The American Society of Apheresis has published guidelines for the use of TPE for several distinct conditions associated with TMA. This list is not comprehensive and the use of TPE for other causes of TMA may be considered if the mechanism of the underlying disease process provides a clear rationale for this intervention.
Learning Objective
To acquire information and clinical tools to augment clinical judgement regarding the use of TPE in the setting of TMA
Introduction
Thrombotic microangiopathy (TMA) is a term that describes the pathological findings of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and microvascular thrombi. The TMA process begins with a pathological insult to endothelial cells, leading to the formation of fibrin- and platelet-rich thrombi in the microcirculation. Platelets are consumed in thrombi, and the shear stress of partially obstructed vessels results in fragmentation of erythrocytes (schistocytes), causing thrombocytopenia and anemia, respectively.1 TMA occurs in multiple clinical settings and the instigating pathological processes differ depending on the associated medical condition. It is essential that the underlying cause of TMA be identified quickly because optimal treatment varies considerably based on diagnosis.2,3
There are numerous diagnostic challenges associated with TMA. First, there is considerable overlap in presenting clinical and laboratory features of TMA-associated diseases. This is exemplified by the general designation of “TTP/HUS” (thrombotic thrombocytopenic purpura/hemolytic uremic syndrome) for many cases of TMA even though the entities have distinct pathologic processes and different optimal strategies for therapy. With a better understanding of the pathophysiology, “TTP/HUS” has essentially vanished from the literature, appropriately, and further use of this term should be discouraged. Second, a considerable amount of diagnostic testing (eg, ADAMTS13 activity and complement mutations) must be performed by specialized reference laboratories and results may not be available for days to weeks after the initial presentation of TMA. Finally, there are cases of TMA for which no underlying pathologic insult can be identified despite extensive testing.
The term “apheresis” describes a variety of procedures for processing blood that allows for the removal or manipulation of a component in a manner that allows for the remaining blood elements to be returned to the patient. Therapeutic plasma exchange (TPE) is a specific apheresis procedure for removing and discarding processed plasma that allows for the return of all cellular blood components back to the patient, along with a replacement fluid to compensate for the discarded plasma. TPE is a relatively safe procedure with minimal associated mild and transient side effects. Rare cases of significant morbidity and mortality in the setting of TPE have been reported, but most of these instances were due to the patient's underlying medical condition or the risks associated with central line placement.4,5
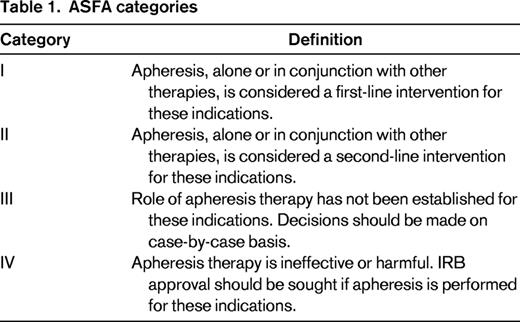
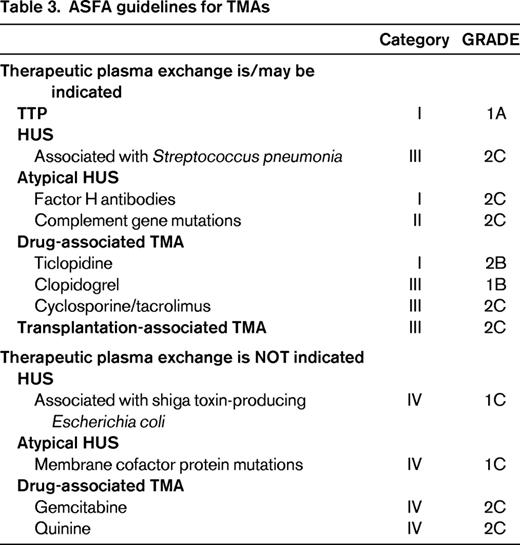
TPE is commonly requested when a patient is discovered to have TMA, often before the underlying cause is elucidated. In many clinical settings, this request is appropriate, but there are other clinical scenarios for which TPE has little to no benefit or may actually be harmful to the patient either directly or indirectly by substituting for a more efficacious therapy. The American Society for Apheresis (ASFA) Journal of Clinical Apheresis Special Writing Committee routinely reviews the published medical literature for all apheresis indications and develops treatment recommendations based on careful scrutiny of the available data.6 The committee assigns a “category” to each indication that designates a recommendation for (or against) apheresis treatment with an associated GRADE based on the Grading of Recommendations Assessment, Development and Evaluation system for evaluating the quality of evidence in the published literature (Tables 1, 2). The most recent ASFA apheresis guidelines, the 6th edition published in 2013, provide recommendations on the use of TPE for 5 specific or general TMA categories: TTP, HUS, atypical HUS (aHUS), drug-associated TMA, and transplant-associated TMA (TA-TMA) (Table 3). This review highlights 3 of these TMA categories, focusing on the unique etiologies leading to TMA and the utility of TPE in each setting. There are several other causes of TMA that will not be reviewed due to space limitation and the reader is referred to recent comprehensive reviews on these conditions.2,3,9-11
TTP
TMA due to TTP is rare; the reported incidence is 4 cases per million people.7 Prompt diagnosis and intervention with TPE or other plasma therapy is critical because patients can quickly decompensate and the untreated mortality is 90%.8 Historically, TTP was characterized by the pentad: MAHA, thrombocytopenia, neurologic abnormalities, renal impairment, and fever. In recent years, the criteria have been revised such that TTP should be considered in the setting of MAHA and thrombocytopenia alone, although >60% of patients will present with associated neurological abnormalities, including altered mentation, headache, paralysis, and coma. Renal failure and fever are not prominent features of TTP.2,5,9
The vast majority of TTP cases are acquired and due to an autoantibody against the VWF-cleaving protease, ADAMTS13.10,11 Congenital TTP, also known as Upshaw-Schulman syndrome, accounts for ∼5% of TTP cases and is caused by mutations in the ADAMTS13 gene that result in persistently low levels or activity of the enzyme.12
TTP pathogenesis and diagnosis
VWF is a variably sized, 540–20 000 kDa, multimeric glycoprotein that is an essential mediator of primary hemostasis. Under normal and pathologic conditions, injured vessels release VWF from the Weible-Palade bodies of endothelial cells, which recruits platelets to the site of injury by acting as a tether between the subendothelial matrix (collagen binding) and platelets (glycoprotein Ib-α receptor). Activated platelets also release VWF, resulting in further platelet recruitment and mediation of platelet–platelet interactions through glycoprotein IIb/IIIa.13 High-molecular-weight/ultra-large VWF multimers are very efficient inducers of platelet aggregation and their size must be regulated to prevent thrombosis. VWF length is primarily controlled by the ADAMTS13 enzyme, which cleaves multimers of VWF into smaller fragments with reduced thrombogenic potential.13
Severe deficiency in ADAMTS13 enzyme activity is generally believed to be the underlying cause of both acquired and congenital TTP.10,11 In the absence of ADAMTS13, ultra-large VWF multimers accumulate in the plasma and are activated by unfolding due to sheer forces in the microcirculation that expose platelet-binding sites. Platelet-rich thrombi form in the systemic microcirculation, leading to MAHA, thrombocytopenia, and tissue ischemia.14
In general, a diagnosis of TTP can be made by demonstrating a profoundly low, usually <10%, ADAMTS13 activity in the plasma from patients with clinical features consistent with TTP.5,10,11 This assay can be used to distinguish between TTP and HUS, and ADAMTS13 activity levels <5% are 90% specific for TTP.15 A majority of these patients will also have an identifiable autoantibody directed against the enzyme. If no autoantibody is detected, congenital TTP should be suspected. Some experts in TTP diagnostics believe that mildly reduced or even normal levels of ADAMTS13 are possible in the setting of TTP16 ; however, this idea remains controversial and may be a consequence of laboratory testing errors or misclassification of the TMA due to referral center reporting.5 Other reports of ADAMTS13 deficiency in non-TMA conditions, for example, fulminant hepatic failure and systemic infections, raise questions regarding the diagnostic utility of ADAMTS13 testing and highlight the importance of interpreting the results within the appropriate clinical context.17
Functional assays to measure the activity of ADAMTS13 are based on the detection of cleaved VWF multimers or recombinant VWF peptides in the presence of patient plasma. These tests can also be used to identify inhibitory autoantibodies directed against the enzyme. These assays are subject to analytical error and several external factors should be considered to interpret the results correctly. For example, patient specimens that are collected and tested after the initiation of TPE may show falsely elevated activity due to the passive transfer of exogenous ADAMTS13 enzyme during the exchange. Alternatively, some activity assays may show falsely low activity when the patient has hyperbilirubinemia or high levels of free plasma hemoglobin.18 Other laboratory features consistent with a diagnosis of TTP include: median presenting platelet count and hemoglobin, 10-30 × 109/μL and 8-10 g/dL, respectively; low haptoglobin, elevated lactate dehydrogenase, negative direct Coombs test, and schistocytes on peripheral blood smear.2
Sequencing of the ADAMTS13 gene is used to confirm definitively a diagnosis of congenital TTP. However, a therapeutic trial of a simple plasma infusion can be helpful in differentiating between idiopathic TTP. Almost half of all patients with congenital TTP are asymptomatic until they suffer from a physiologic stress event as adults, for example, pregnancy.19
Treatment of TTP with TPE
Due to the high risk of mortality associated with TTP, treatment with plasma exchange should be initiated before the diagnosis is confirmed with laboratory testing. The ASFA guidelines state that acquired TTP is a category I indication for TPE that is supported by high-quality evidence (grade 1A).6 Daily plasma exchange until platelet normalization is considered standard treatment for TTP and its use has reduced the mortality associated with TTP from 90% to 10%–20%.8,20 During the exchange, 1-1.5 plasma volumes are exchanged with allogeneic donor plasma products (eg, fresh frozen plasma, cryo-poor plasma). The benefits of TPE are 2-fold, autoantibodies against ADAMTS13 are removed and exogenous ADAMTS13 enzyme is infused with donor plasma.
Patients with congenital TTP do not need plasma exchange, unless they have volume sensitive comorbidities, because these patients do not have autoantibodies to ADAMTS13. These patients can be managed with scheduled plasma infusions to replace the ADAMTS13 enzyme.6
HUS
HUS is a TMA that is characterized by MAHA, thrombocytopenia, and acute renal failure. The most common cause of HUS is an infection with the Shiga-like toxin-producing bacteria, E. coli O157:H7 (STEC-HUS) or related serotypes. In this setting, HUS typically occurs 2-10 days after a prodrome of bloody diarrhea. The incidence of STEC-HUS is reported to be 2 per 100,000 in adults and 6.1 per 100,000 in children <5 years of age.21 The percentage of STEC infections that result in HUS vary considerably and range from a low as 3% in sporadic cases to as high as 20%, as reported in the 2011 outbreak in Northern Europe.21,22 Approximately 50% of patients with STEC-HUS require dialysis and 25% of patients are noted to have central nervous system abnormalities (eg, seizure, stroke, and coma).21 The primary distinguishing element to differentiate TTP from HUS is the presence of oliguric/anuric renal failure in HUS.2
A rare form of HUS triggered by neuraminidase-producing Streptococcus pneumoniae (p-HUS) occurs primarily in children under the age of 2 years and accounts for ∼5% of HUS cases in this population. Mortality during the acute phase of p-HUS is 25%.23
HUS pathogenesis and diagnosis
After ingestion of STEC (or other implicated pathogens), the bacteria colonize the intestinal mucosa by adhering to gut epithelial cells and release shiga-like toxins (Stxs) that translocate across the gut mucosa and enter the bloodstream. Free Stxs have not been detected in the sera of HUS patients; however, neutrophils can bind Stxs and these cells may serve as a delivery vehicle for the toxins.24 Stxs induce inflammatory responses in vascular endothelium, which ultimately lead to generalized microvascular thrombi. The kidney is particularly vulnerable to the effects of Stxs because tubular epithelial and mesangial cells are susceptible to the cytotoxic effects of the toxin.1
The underlying pathologic process in p-HUS is due to neuraminidase production by the bacteria. Neuraminidase cleaves N-acetylneuraminic acid from glycoproteins on cell membranes, in particular RBCs and glomerular cells. Cleavage of this glycoprotein exposes a cryptic epitope known as the T-antigen. Anti-T IgM antibodies are naturally occurring antibodies found in human plasma.25 It is speculated that anti-T antibodies bind to exposed T-antigens on glomerular cells, activating complement and the TMA process.
Diagnosis of HUS depends on the detection of the offending pathogen in culture. If STEC-HUS is suspected, stool cultures should be collected promptly because the positive detection rate is high, >90%, when tested in the week after infection.26 If p-HUS is suspected, culture of body fluids is recommended. Renal failure is present in all patients with HUS and reflected by elevated serum creatinine, low glomerular filtration rates, hematuria, and proteinuria. MAHA and elevated LDH are also noted, however, the degree of thrombocytopenia is typically less that that found with TTP.5,9 The direct Coombs test may be positive in patients with p-HUS due to anti-T binding to newly exposed T-antigens on the RBC membrane.25 In general, ADAMTS13 levels are normal or slightly decreased in the setting of HUS. However, isolated cases of profoundly low ADAMTS13 activity have been reported in STEC-HUS.27 During the recent 2011 outbreak in Northern Europe, 2 of 6 patients with STEC-HUS showed ADAMTS13 activity <5%, although a relapse of “HUS” in 1 of the 2 patients suggests that this patient actually had TTP.28
Treatment of HUS with TPE
The ASFA classifies STEC-HUS as category IV, meaning that TPE is not indicated due to lack of efficacy or potential for harm. This is a strong recommendation based on the current evidence; however, this recommendation may change with future studies.6 At the beginning of the 2011 European outbreak of E. coli O140.H4, the German Society of Nephrology recommended the use of TPE in cases of HUS, particularly those with severe renal and neurologic impairment. A multicenter, retrospective, case-control study of 298 adults in northern Germany with HUS found that TPE was of no benefit in this population and that patients who received prolonged courses of TPE had worse outcomes than those who had limited TPE (3-5 sessions).21 A second, single-center evaluation of 130 patients, 108 of whom received TPE, used platelet counts to determine the efficacy of TPE in the 2011 outbreak of STEC-HUS. These investigators report that 75% of patients did not respond to TPE with respect to improved platelet counts and most patients continued to show renal and neurological declines throughout the course of TPE.29 Although the majority of published evidence does not support a role for TPE in STEC-HUS, there are occasional reports of beneficial responses.6 In general, supportive therapy including dialysis is recommended as the standard treatment for STEC-HUS.
In cases of p-HUS, TPE may be beneficial due to removal of anti-T antibodies and bacterial neuraminidase. One important consideration is the replacement fluid. Typically, plasma products are used in the setting of TMA, but plasma contains naturally occurring anti-T antibodies that could theoretically exacerbate the pathologic process. The recommended replacement fluid in this setting is 5% albumin. The ASFA guidelines list p-HUS as category III with grade 2C evidence and recommend that decisions regarding the duration of TPE be made on an individualized basis depending on response.6
aHUS
aHUS is caused mainly by defects in the regulation of the alternative complement pathway. Historically, the designation of aHUS was applied to the rare cases of HUS for which there was no apparent infection with the classic pathogens known to be implicated in HUS. These cases account for ∼10% of HUS, with annual incidences of 2 per million in adults and 3.3 per million in children <18 years of age.22 aHUS tends to be chronic condition, unlike HUS, which is a one-time event. Most patients will present with the classic HUS triad of MAHA, thrombocytopenia, and renal failure, but a minority of patients will have mild or fluctuating anemia/thrombocytopenia and may have no overt evidence of renal disease at diagnosis.21 aHUS is associated with significant morbidity and mortality: 65% of patients have end-stage renal disease or die within the first year of presentation.30 Genetic or acquired defects that lead to uncontrolled activation of the alternative complement pathway have been found in 60% of aHUS cases.30
HUS pathogenesis and diagnosis
Glomerular endothelial cells are particularly vulnerable to complement dysregulation and the TMA-inducing microvascular lesions in aHUS are primarily located in the kidney.21 More than 120 reported mutations in the alternative complement pathway proteins have been identified. Complement factor H (CFH) is the most important plasma regulator of the alternative pathway. Mutations causing loss of function or deficiency in CFH account for 20%–30% of aHUS cases.30 There are also acquired deficiencies of CFH due to the generation of anti-CFH autoantibodies. Loss of function mutations have been reported in complement factor I (CFI) and membrane cofactor protein (MCP).31 Mutations in complement factor B (CFB) and in complement component 3 (C3) resulting in gain of function account for other cases of aHUS.32 Detailed descriptions of the numerous complement mutations associated with aHUS are beyond the scope of this review, but several excellent comprehensive reviews have been published elsewhere.22
Variable penetrance has been reported in ∼50% of people identified with mutations in CFH, CFI, MCP, CFB, and C3. Therefore, aHUS is likely triggered by a precipitating event in genetically vulnerable people. Precedent upper respiratory or gastrointestinal infections have been reported in more than half of patients with aHUS.30 Interestingly, complement mutations have also been found in patients with other TMA diagnoses, including p-HUS and preeclampsia.33,34 Jodele et al performed complement genetic testing on 6 consecutive pediatric patients who developed TA-TMA after autologous or allogenetic hematopoietic stem cell transplantation.35 These investigators found deletions in the complement factor H-related genes 3 and 1 (CFH3-CFH1) in 5 of the 6 children with TA-TMA.
The diagnosis of aHUS is often not considered until after TTP and HUS have been ruled out. Laboratory tests for complement should include serum levels of C3, C4, CFH, and CFI, in addition to anti-CFH autoantibodies. Although C3 levels are often reduced in patients with complement pathway defects, normal levels do not rule out aHUS.36 MCP is a membrane-bound protein and deficiencies may be observed using fluorescence-activated cell sorter analysis of peripheral blood mononuclear cells. Specialized reference laboratories are required for the detection of mutations in complement genes and for other sophisticated complement factor analysis. Given the variable degrees of TMA and renal failure in patients with aHUS, laboratory values for hemoglobin, platelet counts, LDH, and creatinine may not be as severely abnormal when compared to TTP and HUS. ADAMTS13 activity is reportedly normal in TMA associated with aHUS.37
Treatment of aHUS with TPE
The use of TPE in aHUS is based on the rationale that this therapy will remove the defective complement proteins and autoantibodies against CFH in addition to providing normal exogenous complement factors when allogeneic donor plasma is used as the replacement fluid for the exchange. ASFA guidelines indicate that TPE is a first-line therapy for patients with anti-CFH autoantibodies (category I, grade 2C) and a second-line therapy for other complement mutations (category II, grade 2C). The exception is mutations in MCP; this protein is bound to cellular membranes and is not found circulating in plasma. TPE has not been shown to benefit patients with MCP mutations and therefore this indication is listed as a category IV.6
The response to TPE is similar in aHUS patients with and without identified complement mutations and therefore treatment with TPE should be started immediately and continue pending response or the results of complement testing. Hematologic remission of aHUS is defined as normalized platelet counts for >14 days with no evidence of ongoing hemolysis.6 Treatment with eculizumab, a humanized monoclonal antibody that targets complement component 5 (C5) that is approved by the U.S. Food & Drug Administration for use in aHUS, should be strongly considered in cases of aHUS due to the published evidence showing clinical benefit.38
TPE for other TMA-associated conditions
TMA is associated with a disparate group of clinical conditions that cause endothelial damage in the microcirculation through a variety of mechanisms (Table 4). Targeting the underlying insult is key to resolving TMA, and the use of TPE should be considered based on whether there is plausible evidence that the abnormality can be meaningfully corrected with this intervention.
Conclusion
Elucidation of the pathological processes of TMA, such as ADAMTS13 deficiency in TTP or complement mutations in aHUS, has led to improved outcomes in patients through enhanced diagnostic accuracy and rationally designed targeted therapy. Given the risks of morbidity and mortality and the potential benefit of TPE in several clinical settings associated with TMA, immediate initiation of TPE should be considered while the diagnostic workup is in progress. If the underlying cause of TMA is determined to be a medical condition that does not respond to TPE or is known to have superior alternative therapies, TPE should be discontinued.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Jill Adamski MD, PhD, Department of Laboratory Medicine and Pathology, Mayo Clinic Arizona, 5777 E Mayo Blvd, Phoenix, AZ 85054. Phone: 480-342-2606; Fax: 480-342-3658; e-mail: adamski.jill@mayo.edu.