Abstract
The primary function of red blood cells (RBCs) is to deliver oxygen from the lungs to tissues. Tissue hypoxia occurs when the oxygen-carrying capacity of RBCs is compromised due primarily to 3 causes: (1) a reduction in circulating RBC mass, (2) an increase in circulating RBC mass, or (3) abnormal hemoglobin (Hb) that either does not sufficiently release oxygen to tissues (high-oxygen-affinity hemoglobin) or occludes the microvasculature due to deformed RBCs (sickled RBCs). To improve oxygenation in patients with reduced or increased RBC mass, RBC administration (simple transfusion) or RBC removal (RBC depletion) is performed, respectively. However, for patients with abnormal Hb, RBCs containing abnormal Hb are removed and replaced by healthy volunteer donor RBCs by red cell exchange (RCE). RCE can be performed by manual exchange or by automated exchange using a blood cell separator (erythrocytapheresis). In this review, indications for RCE in sickle cell disease using the evidence-based American Society for Apheresis categories1 are presented and the rationale for RCE in each disorder are discussed. Simple transfusion versus RCE and manual RCE versus automated RCE are compared. Finally, this review briefly presents some of the challenges of performing erythrocytapheresis in small children and discusses various choices for central venous access during RCE.2
Learning Objectives
To describe indications for RCE
To define goals for RCE for stroke prophylaxis in sickle cell disease
To discuss benefits versus risks of automated RCE compared with simple transfusion and manual RCE.
Introduction
The primary goal of therapeutic red cell exchange (RCE) is to remove abnormal or excess RBCs from a patient. The 3 indications for RCE are: (1) polycythemia or erythrocytosis, (2) hemochromatosis, and (3) the presence of abnormal RBCs due to intrinsic RBC disorders or acquired RBC disorders resulting from systemic causes such as infections, drugs, or chemicals. For the first 2 indications, RCE is performed to deplete RBCs or the iron store with non-RBC replacemenst. The hallmark of erythrocytosis and polycythemia is increased hematocrit (Hct) with hyperviscosity of whole blood with altered blood rheology, resulting in hypoxia and increased thrombosis.3 Manual phlebotomy is still the standard treatment in polycythemia/erythrocytosis and hemochromatosis. The first prospective randomized trial in hereditary hemochromatosis to achieve the target serum ferritin level ≤50 μg/L showed that erythrocytapheresis is highly effective in reducing iron overload and lowering the total number of procedures by at least 50% compared with phlebotomy, and it also shortens treatment duration.4 For the third indication, RCE is performed to remove abnormal RBCs and replace them with donor RBCs.
RCE can be used as acute or chronic transfusion therapy; acute RCE is used to treat severe or life-threatening complications of RBC disorders and chronic RCE is used to prevent either new or recurrent adverse events or progression of preexisting organ dysfunction. To date, the advantages of RCE, especially compared with simple transfusion, and the relative benefit of manual versus automated RCE, have rarely been documented in prospective randomized controlled trials. In this review, RCE refers to both manual and automated RCE; however, when only automated RCE is used in a study, this will be referred to as erythrocytapheresis. Readers are referred to excellent reviews on RCE.5-8
Indications for RCE
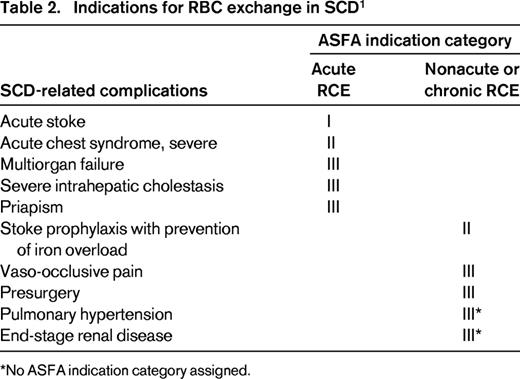
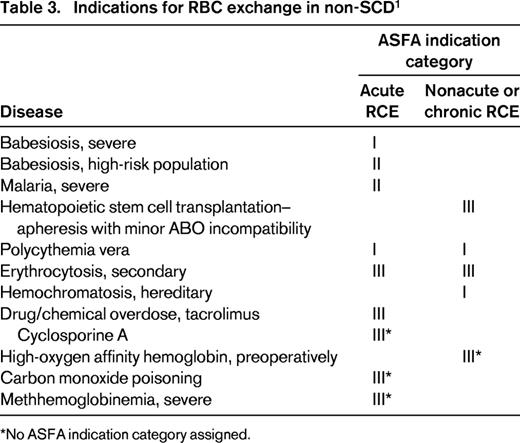
RCE is used exclusively in treating complications of sickle cell disease (SCD), but is rarely used in non-SCD disorders. In this review, RCE as acute or chronic transfusion therapy in only SCD-related complications is presented and the rationale for RCE in a specific complication entity will be discussed. The American Society for Apheresis (ASFA) classifies indications for therapeutic apheresis into 4 categories on the basis of evidence.9 The 4 ASFA categories are defined in Table 1. Table 2 summarizes the indications for RCE in SCD. Indications for RCE in non-SCD disorders are listed in Table 3 using the ASFA indication categories without further review.
In SCD, sickle hemoglobin (HbS) polymerizes upon deoxygenation, causing RBCs to become rigid, adherent, and deformed, leading to microvasculature occlusion, which results in tissue hypoxia and infarction. In addition to impairment of RBC rheology due to rigid RBCs, many other factors are responsible for the pathophysiology of SCD, such as dysfunction of the vascular endothelium, specifically, hemolysis-associated low arginine and nitric oxide bioavailability, increased inflammatory cytokines, and hypercoagulable state.10 Despite much progress in understanding the pathophysiology of SCD and innovations in medical treatments, to date, no therapy is completely effective for the treatment and prevention of complications of SCD. At the present time, the standard common practice for treating SCD-related complications is transfusions of donor RBCs to increase the oxygen-carrying capacity of the blood by reducing HbS concentration and increasing the Hb level.
For acute complications of SCD, the goal of transfusion therapy is to reduce the posttransfusion HbS level to <30%; for chronic complications, the goal is to maintain the pretransfusion HbS level at <30%-50% while maintaining the Hb level at ∼10 g/dL. Rapid lowering of HbS levels can only be achieved by acute RCE.
Category I indication
Acute stroke (cerebrovascular accident)
Cerebrovascular accident is one of the most devastating complications of SCD. Stroke occurs in both children and adults and, without preventive therapy, ∼11% of patients experience stroke by 20 years of age.11 In the absence of transfusion therapy, recurrent strokes occur in ∼2/3 of SCD patients after the first cerebrovascular accident.12
Current practice for treating acute stroke involves acute transfusion to maintain HbS <30%. Acute simple transfusion raises the Hb level, which improves oxygen delivery to the brain, but will not sufficiently reduce the HbS level without raising the Hb level much higher than 10 g/dL, which is associated with hyperviscosity. A retrospective cohort study of 137 children with SCD and first strokes showed that children receiving simple transfusion at the time of stroke presentation had a 5-fold greater relative risk of having a second stroke than those receiving RCE.13 In that study, RCE (both manual and automated) was the most common initial treatment for overt stroke. There have been no prospective randomized trials comparing different methods of transfusion as an initial treatment for stroke.
Compared with simple transfusion, acute erythrocytapheresis has several distinctive advantages in patients with SCD and overt stroke: (1) a low Hb level can be raised rapidly without circulatory volume overload, (2) the HbS concentration can be lowered rapidly to a predetermined level without raising the Hb level >10 g/dL, and (3) erythrocytapheresis can be performed safely under isovolemia. Despite the lack of clinical trials, emergent RCE, especially erythrocytapheresis, is preferred over simple transfusion for treatment of acute stroke because treatment goals for postexchange Hct of ∼30% (not higher than 36%) and HbS <30% can be achieved rapidly.
Category II indications
Acute chest syndrome
Acute chest syndrome (ACS) is defined by the National Acute Chest Study Group as a new infiltrate on chest X-rays accompanied by at least one of the following: chest pain; fever >38.5°C; and respiratory findings such as tachypnea, cough, or wheezing. These symptoms can rapidly progress to respiratory distress, leading to severe hypoxia, worsening anemia, and sometimes cardiopulmonary failure. ACS is the most common cause of death in adults with SCD and the second most common cause of hospitalization. The management of ACS includes antibiotics, respiratory therapy, and RBC transfusion.
In a retrospective study of 32 episode of ACS in 32 children, all 23 children who received transfusion within 24 hours after hospital admission had improved outcomes, whereas 5 of the 9 children who did not receive transfusions deteriorated.14 In a prospective study of 40 episodes of ACS in 36 children with SCD, >65% of episodes were treated with transfusion (simple transfusion, RCE, and simple transfusion followed by whole blood exchange). The transfusions significantly improved oxygenation, but there was no significant difference in clinical manifestations and duration of hospital stay between the transfused and nontransfused groups.15
In a multicenter prospective study of 671 episodes of ACS in 538 SCD patients in the United States, 72% of the patients were transfused for a worsening clinical course, with both simple transfusion and RCE associated with improved oxygenation, which suggested that limited transfusions alleviate organ dysfunction.16 In contrast, in a monocentric series of 107 episodes in 77 adult patients from France, 50 episodes were treated with transfusions including simple transfusion and RCE.17 There were 5 deaths in the transfused group and none in the nontransfused group. At this center, transfusion was administered only in the most severe cases, which were defined as patients with early acute respiratory failure or those with mild respiratory distress that worsened after 3 days. It is not clear whether deaths in the transfused group were attributed to more severity of illness than those who were not transfused. The investigators concluded that restricting transfusions to the most severe ACS cases does not seem to increase the mortality rate, so transfusions should be restricted to the more severely affected cases.
The effects of chronic transfusion therapy on the prevention of new and recurrent episodes of ACS in 27 children with SCD were reviewed.18 Transfusion significantly reduced the incidence of ACS events, but did not significantly decrease their severity. Comparing simple transfusion with RCE in 40 adults with ACS using a retrospective review, investigators found no difference in patient outcome between RCE and simple transfusion.19 A retrospective study of 53 episodes of ACS in 44 children who underwent erythrocytapheresis for worsening respiratory function demonstrated that clinical respiratory scores were improved within 24 hours after exchange.20
These studies indicated that early transfusions appear to be beneficial for patients with severe clinical manifestations or high risk for complications, such as a history of cardiac disease and severe pain in the extremities at presentation. Furthermore, prompt RCE over simple transfusion should be considered in selective patients with ACS including those with: (1) rapidly worsening respiratory distress, (2) worsening chest X-ray, (3) worsening hypoxemia requiring increased oxygen support or mechanical ventilation, (4) failure to improve with simple transfusion, or (5) concurrent neurologic deficit or pulmonary embolism.
To date, there have been no prospective randomized controlled trials to demonstrate the effectiveness of transfusion over standard therapy or to compare simple transfusion with RCE in patients with ACS. Efforts should be made to better delineate the criteria for transfusions and to demonstrate the effectiveness of transfusion over standard therapy in patients with ACS, in particular comparing simple transfusion with erythrocytapheresis. Several studies have demonstrated that chronic transfusion therapy for stroke prevention or other SCD-related complications reduced recurrent ACS and as painful crises.16,21-23 The effectiveness of chronic transfusion therapy should be compared prospectively with that of hydroxyurea and stem cell transplantation.
Stroke prophylaxis with iron overload prevention
Chronic transfusion therapy to maintain HbS at <30% remains the mainstay of therapy for recurrent (secondary) stroke prevention. The Stroke Prevention Trial in SCD (STOP I) demonstrated that maintaining HbS at <30% reduced the annual incidence of first (primary) stroke in children with an abnormal TCD velocity by >90% compared with those who received standard care.23 STOP II indicated that transfusions could not be safely discontinued to prevent first stroke.24 In these trials, the methods of transfusion included simple transfusion, RCE, or a combination of both. Currently, the standard care for prevention of primary and secondary stroke is to maintain the target HbS at <30% by indefinite long-term transfusion regardless of the method of transfusion. In selected cases in which there is no progression of vasculopathy and/or recurrent neurologic symptoms, a target HbS of <50% may be as effective as HbS <30% in preventing recurrent stroke.25
Long-term transfusion therapy is associated with potentially serious complications, among them transfusional iron overload. Both oral and parenteral iron chelation are effective in treating iron overload, but treatment failure is common due to poor compliance, drug toxicity, and/or intolerability.
In an effort to prevent transfusional iron overload, chronic transfusion therapy has been modified by raising the target HbS level to <50% from <30% or by replacing simple transfusion with manual RCE in selected patients. In the mid-1980s, when the blood cell separator for erythrocytapheresis became available, manual RCE was switched to automated RCE (erythrocytapheresis). Kim et al reported that long-term erythrocytapheresis with pre-exchange target HbS <50% prevented or markedly reduced transfusional iron overload in 14 patients with SCD.26 Subsequently, several centers confirmed this finding.27-30 However, the main problem with erythrocytapheresis are the significantly increased blood requirements compared with simple transfusion. To reduce blood requirements, Kim et al modified the standard erythrocytapheresis procedure by incorporating RBC depletion with isovolemic hemodilution into the automated procedure.26
Although there have been no controlled trials comparing chronic simple transfusion with chronic RCE, numerous studies from case reports and series support the efficacy of chronic RCE in preventing iron overload, particularly erythrocytapheresis over manual RCE, which is an acceptable therapy on an adjunctive basis for prevention of both recurrent stroke and transfusional iron overload (Table 2).
Category III indications
Vaso-occlusive pain
Pain is the most common manifestation of vaso-occlusive events in patients with SCD, requiring frequent emergency department visits and hospitalizations. Molecular and vascular/neurobiological studies in human and animal models have shown that, in addition to vaso-occlusion, multiple factors are responsible for painful episodes. Despite much progress in understanding the pathophysiology of SCD and innovations in medical treatments, to date, no therapy is completely effective for the treatment and prevention of acute pain or recurring/relapsing chronic pain. Readers are referred to excellent reviews on the pathophysiology of, and novel therapies for, SCD-related pain.31-33
Prevention of recurrent or chronic pain.
Kalff et al evaluated the impact of a chronic erythrocytapheresis program on the incidence and types of acute and chronic complications of SCD and transfusions in 12 adult patients with various complications of SCD.21 The median duration of follow-up was 70 months (range 8-119 mo). The mean HbS levels immediately before and after each erythrocytapheresis treatment were <59% and <33%, respectively. In this group, the median number of painful crises per patient requiring admission in the 5 years before erythrocytapheresis was 8, whereas a total of 11 painful crises occurred over 415 months of follow-up. No patient developed stroke, multiorgan crises, or end-organ dysfunction while on a chronic erythrocytapheresis program. Subsequent correspondence by Driss et al described similar beneficial effects of regular erythrocytapheresis in 43 patients with SCD.34 In this group, the mean HbS levels immediately before and after erythrocytapheresis were 48% and 20%, respectively. Those investigators noticed that erythrocytapheresis was either ineffective in severe priapism or resulted in inconsistent outcomes for those with leg ulcers. These studies demonstrate that, with chronic erythrocytapheresis therapy, maintaining HbS levels at <50%-60% is effective in reducing sickle-related acute and chronic complications, thus reducing hospitalization and morbidity. In fact, painful crises were precipitated by stopping prophylactic RCE.35
Preoperative management
The standard preoperative transfusion protocol for patients with SCD is to increase the Hb level to 10 g/dL by simple transfusion.36,37 However, for patients with an Hb level >10 g/L, such as those with SCD-SC or SCD-S-beta thalassemia, RCE is the preferred method of transfusion because it effectively reduces HbS to <30% while maintaining the Hb level at 10 g/dL. RCE, particularly using erythrocytapheresis if available, with the target HbS <30% should be considered instead of simple transfusion in patients with significant comorbidities and/or undergoing major procedures such as open heart surgery or retinal surgery even though their Hb level is low.
Priapism
Priapism is defined as painful, persistent, and unwanted penile erection due to vaso-occlusion. The incidence of priapism is 35%.38 There are 2 types: prolonged (lasting >3 hours) and stuttering (lasting <3 hours). Conventional treatments include intravenous hydration, analgesics, intracavernosal aspiration, and instillation of an alpha-agonist. There have been no controlled trials to compare transfusion or RCE with conventional therapy in the treatment of priapism. A retrospective review including 42 case reports comparing transfusion with conventional therapy demonstrated that adverse neurologic sequelae from transfusion were found in 9 of 26 transfused cases, with outcomes ranging from complete resolution to severe residual deficits. The investigators concluded that routine use of transfusion in the treatment of priapism was not supported.39
In a small number of case reports and series, erythrocytapheresis has been associated with variable clinical response, ranging from resolution within 6-8 hours40 to no resolution.41 Some sickle cell patients with priapism who undergo RCE have also experienced a serious neurological complication known as Aspen's syndrome (which is an acronym for association of sickle cell disease, priapism, exchange transfusion, and neurological events).42 However, all of these cases had significantly increased postexchange Hct levels. Despite conflicting outcomes with RCE, erythrocytapheresis with the goal of reducing the HbS level to <30% and achieving a postexchange Hct of ∼30% may be considered if early intervention with irrigation fails.43
Acute multiorgan failure syndrome
Acute multiorgan failure syndrome is defined as acute failure of at least 2 of the following organs: lung, liver, or kidney. Acute multiorgan failure syndrome is a severe, life-threatening complication of SCD, which occurs rarely and is often associated with severe pain episodes in patients with relatively high Hb values in a steady state. Treatment requires prompt and aggressive transfusion therapy, which can potentially reverse organ failure and reduce mortality. In a retrospective review of 17 episodes in 14 patients, 16 episodes were associated with rapid recovery of organ failure occurring within 24 hours of transfusion regardless of the method of transfusion.44 In this review, death rates (1/9 cases vs 0/8 cases) were similar between RCE and simple transfusion; however, the mean times for discharge (7 vs 15 days) and for complete organ recovery (2 vs 3-6 months) were shorter with RCE.
Intrahepatic cholestasis
Sickle cell hepatopathy or intrahepatic cholestasis is an uncommon complication of SCD, but is associated with fulminant hepatic failure and a very high mortality. There are 2 types of intrahepatic cholestasis, mild and severe/fatal.45 In the severe type, the total serum bilirubin level is usually >30 mg/dL. In a retrospective review of 7 patients from a single center and 37 patients from the literature review, 50% of patients had the severe type of disease with a mean bilirubin level of 76.8 mg/dL (range 14-273).46 In this group, the mortality rate was >60%; 2 of 9 patients who received RCE died, whereas 12 of 13 patients who did not receive RCE, but received treatment including simple transfusion, died. This study and other case reports indicate that prompt RCE is the first line of therapy to prevent fatal outcomes and that RCE is markedly more effective compared with simple transfusion in reducing mortality.
Uncategorized indications
Not all diseases or disorders are included for categorization by the ASFA Writing Committee. The following SCD-related complications are uncategorized indications for RCE in which the role of RCE has not been determined definitively. The decision to perform RCE in a specific clinical entity can be made by the individualized decision maker under the Category III indication.
Pulmonary hypertension
The Sickle Cell Pulmonary Hypertension Screening Study documented that the frequency of pulmonary arterial hypertension (PAH) in adults with SCD is 32%.47 PAH and chronic lung disease are 2 of the most common causes of mortality in SCD. Although there is a lack of clinical trials or published data (other than an abstract) on the use of chronic RCE in patients with SCD and PAH, chronic transfusion therapy should be considered in these cases, especially when hydroxyurea fails to reduce excessive hemolysis, the rate of ACS, or painful crises.48 At our center, a few patients with SCD and PAH have been maintained on chronic erythrocytapheresis for several years, all without further deterioration of pulmonary function (personal observation). Of transfusion methods, erythrocytapheresis is the choice of transfusion in patients with SCD and PAH because automated exchange effectively reduces the HbS level without circulatory overload and prevents transfusional iron overload.
End-stage renal disease/renal transplantation
Patients with SCD develop chronic sickle cell nephropathy due to recurrent intrarenal sickling with proteinuria and a progressive decline in the glomerular filtration rate, which may lead to end-stage renal disease requiring dialysis or transplantation in up to 30% of adults.49 There is little evidence of the benefits of long-term blood transfusions for the prevention of renal complications. As renal function declines, erythropoietin levels also decline, and these patients may require substantially higher doses of erythropoietin. If erythropoietin is ineffective, transfusions can be given; however, care must be taken to avoid volume overload and raising the Hct level to >30% to reduce the risk of triggering vaso-occlusive crises.
Erythrocytapheresis may offer benefits over chronic simple transfusion because it prevents volume and iron overloads. After renal transplantation, Hb levels significantly increase, with a corresponding increase in the number of painful crises due to an increase in whole blood viscosity.50 These patients may benefit from RCE or even from periodic phlebotomy, particularly when Hb levels are high. In patients with SCD, acute renal failure can occur as a component of acute multiorgan failure.
Adverse effects of RCE therapy
Compared with simple transfusion, the adverse effects of RCE primarily result from the use of a large volume of blood and the inherent problems associated with the use of equipment and the possible need for central venous access. The risks include hypovolemia or circulatory overload, citrate toxicity with hypocalcemia, infection, and bruising or hematoma at the site of catheter placement. This review only focuses on alloimmunization to RBC antigens.
RBC alloimmunization
The overall incidence of alloimmunization in patients with SCD ranges from 18% to 76%. Using the RBCs with limited phenotype matched for C, E, and K antigens, the incidence is significantly reduced to 5%–14.5%.51 The estimated incidence of delayed transfusion reactions from small case series was ∼5% in transfused patients with SCD.52 Many patients also develop multiple alloantibodies that can complicate serologic evaluation. Strategies to reduce alloimmunization risk in SCD include use of RBCs with limited and extended phenotypes matched and RBC units collected from African-American donors or limited number of dedicated donors. However, these approaches make for more difficult inventory management and procurement of the desired RBC units in time for transfusion.53 More recently, Chou, et al demonstrated that use of RBC units provided by African-American donors with negative C, E, and K antigens did not reduce Rh alloimmunization, with approximately 30% of unexpected Rh antibodies being associated with laboratory evidence of delayed transfusion reactions.54 Future studies are warranted to determine whether selection of RBC units for patients on the basis of Rh genotyping of patients and donors will reduce and/or prevent Rh alloimmunization.
Erythrocytapheresis requires significantly more blood compared with simple transfusion. Increased blood donor exposure has been associated with higher alloantibody formation. Wahl et al evaluated the effects of chronic transfusion in 45 children with SCD on alloimmunization rates by retrospective review.55 The overall rate of alloantibody formation per 100 units was 0.055 alloantibodies, whereas those in the simple transfusion and erythrocytapheresis groups (23 vs 22) were 0.143 and 0.013, respectively (P = .003), despite the latter group receiving significantly more blood (152.2 units/patient vs 338.5 units/patient). Contrary to the predictions of increased alloimmunization with erythrocytapheresis, limited studies suggest that the risk of higher alloantibody formation with erythrocytapheresis may not be a great concern for its use as a chronic transfusion therapy.
Procedural guidelines for erythrocytapheresis
Automated apheresis instruments require a fixed volume of blood to fill the disposable set and for processing. This volume refers to the extracorporeal volume (ECV). ECV is dependent on both the type of instrument used and the type of apheresis procedure. Because the ECV of each instrument is fixed, it will likely represent a larger fraction of total blood volume for a child than for an adult. In general, if the ECV is >15% of the total blood volume or if the patient is hemodynamically unstable or has severe anemia, a RBC prime may be indicated.2 Apheresis equipment will calculate the replacement packed RBC volume to achieve the desired postexchange target Hct level and fraction (ie, percentage) of a patient's RBC remaining in circulation (FCR). Similarly, these instruments calculate the volume of blood removal necessary to achieve the target Hct level and replacement fluid volume for RBC depletion.
For acute erythrocytapheresis, FCR should be calculated to reduce the level of abnormal RBCs to the desired level. In general, a single 2-volume RCE can reduce the FCR to ∼<15%-20% of the original. For acute erythrocytapheresis in SCD, the procedure should aim for a postexchange Hct at ∼30% (<36% to avoid hyperviscosity) and FCR at <30%. In recently transfused patients, one can calculate the FCR by dividing the desired HbS level by the pre-apheresis HbS level and then multiplying by 100. One procedure is typically sufficient to treat acute complications of SCD.
To prevent iron overload in chronic erythrocytapheresis therapy in SCD, postexchange Hct is not raised >27%. However, if the pre-apheresis Hct is >27%, the postexchange Hct should be the same as the initial Hct unless iron overload exists. To reduce replacement RBC volume, erythrocytapheresis is performed using isovolemic hemodilution, which consists of RBC depletion with 0.9% NaCl replacement followed by standard RCE. Replacement RBC should be HbS negative, leukoreduced, if available, and antigen matched for at least C, E, and K antigens.
Vascular access
Adequate vascular access is a prerequisite for a successful apheresis procedure. Every effort should be made to use peripheral veins for an apheresis procedure, especially for a one-time procedure. When peripheral veins of patients are not adequate, a vascular access device must be placed in a central vein for adequate flow. A percutaneous temporary dual-lumen apheresis or hemodialysis catheter should be placed for critically ill patients requiring acute RCE.
For long-term use, a tunneled and cuffed central venous catheter or single- or dual-lumen Vortex ports (Angiodynamics) have both been effectively used in children and adults who require long-term erythrocytapheresis. In general, ports are easier to care for than central venous catheters, only requiring a heparin flush every 3-5 weeks when not in use. Guidelines at our center for the types and sizes of catheter or ports based on the weight of the patient are given in Table 4.2
Future directions and conclusions
With the ready availability of automated cell separators and ease in technical performance, erythrocytapheresis is being used increasingly to treat acute and chronic complications of RBC disorders, particularly in patients with SCD. As opposed to automated RCE, manual RCE is labor intensive, prolonged, and perhaps less safe and efficient than erythrocytapheresis.
In SCD, both transfusion methods (simple transfusion and erythrocytapheresis) offer similar benefits in maintaining target HbS levels for long-term transfusion therapy. Although simple transfusion is available worldwide and is simple to perform, erythrocytapheresis is not universally available, requires experienced personnel to perform, and may require a central venous catheter/port. The distinctive benefits of chronic erythrocytapheresis are prevention of iron overload. In addition, erythrocytapheresis may avoid the risk of circulatory volume alterations and hemodynamic distress, and thus is a safer procedure than other methods of transfusion. However, to date, the advantages and efficacy of RCE have not been substantially documented through clinical trials, especially compared with simple transfusion or manual versus automated RCE. Although it is difficult to compare simple transfusion with erythrocytapheresis as an acute transfusion therapy with a prospective randomized controlled study, due to the severe or life-threatening presentation of some of these RBC disorders and the relative rarity of symptomatic diseases, it may be feasible to conduct prospective trials to compare these 2 chronic transfusion therapies in the future.
Acknowledgments
The author thanks Frederick Kim for editorial assistance and our apheresis staff for their dedication.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Haewon C. Kim, MD, Division of Hematology, Children's Hospital of Philadelphia, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104; Phone: (215)590-2260; Fax: (267)426-0935; email: kimh@email.chop.edu.