Abstract
Antigen-specific immunotherapies have emerged as important components of curative treatment algorithms for many cancers. In acute myeloid leukemia (AML), success has been less obvious. Nonetheless, among the few drugs shown to improve survival in recent randomized trials is the CD33 antibody–drug conjugate gemtuzumab ozogamicin. Significant antileukemic activity is also well documented for radioimmunoconjugates targeting CD33, CD45, or CD66. These therapeutics can intensify conditioning before hematopoietic cell transplantation, but their effect on patient outcomes needs clarification. Emerging data now suggest clinical antileukemic activity of several novel antibodies and perhaps some adoptive T-cell immunotherapies and vaccines. In parallel, numerous other agents targeting a wider variety of antigens are currently being explored. However, the antigenic heterogeneity characteristic of AML is a considerable limitation for all these therapeutics, and many important questions related to the ideal target antigen(s), disease situation in which to use these therapies, most suitable patient populations, exact treatment modalities, and details of supportive care needs remain open. Addressing such questions in upcoming studies will be required to ensure that antigen-directed therapies become an effective tool in AML, a disease for which outcomes with standard “3 + 7”-based chemotherapy have remained unsatisfactory in many patients.
Learning Objectives
To gain an overview of current clinical results with antigen-specific immunotherapies for acute myeloid leukemia
To become familiar with recent advances and evolving treatment concepts in the field of antigen-specific immunotherapy for acute myeloid leukemia and to appreciate the challenges in the clinical translation of these therapeutic strategies
Conceptually, antigen-specific immunotherapies offer a versatile means of eliminating tumor cells with limited nonspecific toxicities. For many human cancers, including some hematological malignancies, such therapies have emerged as important components of curative treatment algorithms.1-3 Success has been more modest for acute myeloid leukemia (AML). Nonetheless, among the few drugs that have shown to improve survival in recent randomized trials is the CD33 antibody–drug conjugate gemtuzumab ozogamicin (GO). The clinical experience with GO provides the strongest glimpse thus far of the potential value of antigen-targeted therapies for AML. Here, we review the results of efforts with antibodies and other antigen-directed passive and active immunotherapies in AML and summarize evolving treatment strategies for this disease.
Target antigens for immunotherapy in AML
The choice of target antigen is of critical importance for the success of any AML immunotherapeutic.4 Ideally, it is displayed homogeneously at high levels on all AML cells including leukemia stem cells (LSCs), has minimal to no expression in normal tissues, plays an important role in AML pathogenesis, is not shed into the circulation, and is sufficiently immunogenic in the case of active immunization strategies. There are many well-characterized antigens in AML, but finding the perfect target(s) is an unresolved challenge. A main obstacle is antigenic heterogeneity across individual leukemia cells within a patient and between patients because of the clonal and genetic diversity of the disease. As a consequence of the latter, AML-specific antigens (eg, those resulting from chromosomal translocations or gene mutations) are confined to particular patient subsets, limiting their general usefulness and necessitating an individualized treatment strategy. Few antigens on AML cells are restricted to normal embryonic and fetal development (“oncofetal antigens”) or immune-privileged tissues (“cancer–testis antigens”). The vast majority of AML cell antigens described to date are “leukemia associated” and thus also found on nonleukemic cells, especially those of hematopoietic origin, including progenitor and stem cell populations. Toxicity attributable to normal counterpart cell targeting, particularly prolonged cytopenias, is a major concern with all AML-associated antigens and an important factor in the consideration of what constitutes a clinically targetable antigen.
Initially, research on antibody-based therapy in AML has primarily focused on the myeloid differentiation antigen CD33 (SIGLEC-3). CD33 is attractive because it is displayed on at least a subset of leukemic blasts in almost all AML patients,5,6 is possibly present on LSCs in some patients,7 and has endocytic properties that enable its use for targeted delivery of cellular toxins. However, CD33 expression is relatively low, with an average of ∼104 molecules per AML blast and varies over 2-log–fold among patients; thus, some leukemia cells express very few CD33 molecules. For the delivery of radionuclides, CD45 and CD66 have served as alternatives to CD33.8 The number of antigens exploited for immunotherapeutic purposes has recently expanded dramatically. Some are chosen primarily because of their overexpression on AML cells relative to normal cells, including Wilms tumor 1 (WT1) and proteinase 3 (PRTN3). Increasingly, targets are selected because they are important for proliferation, survival and/or drug resistance, or other AML cell functions [eg, CXC chemokine receptor 4 (CXCR4), vascular endothelial growth factor (VEGF), or fms-like tyrosine kinase-3 (FLT3; CD135)] or because they are expressed on putative LSCs [eg, CD44, CD47, CD96, CD123, C-type lectin-like molecule-1 (CLL-1), or interleukin-1 (IL-1) receptor accessory protein (IL1RAP)] or on surrounding cells that mediate host immune responses against AML cells [eg, killer inhibitory receptors (KIRs) on natural killer (NK) cells]. Broadly speaking, the goals pursued with any immunotherapeutic are direct targeting of AML cell antigens, boosting the number and activity of immune effector cells, activating tumor antigen-specific immunity, and/or overcoming inhibitory immune suppression.3
Clinical results with unconjugated antibodies
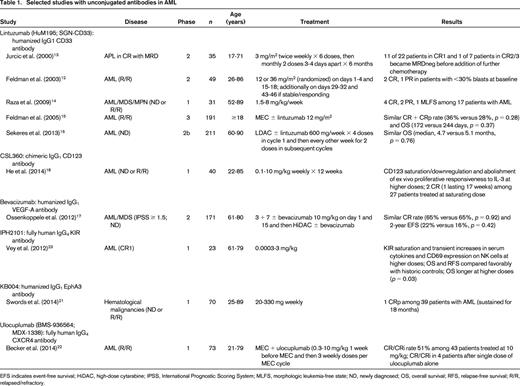
Although many unconjugated antibodies against a variety of antigens have shown efficacy in preclinical AML models, they typically were unsuccessful when investigated in the clinic (Table 1). Among these were antibodies targeting CD15 or CD52 (alemtuzumab), which were given to only a handful of patients.9,10 Most widely tested was the CD33 antibody lintuzumab (HuM195, SGN-CD33), which produced infrequent complete remissions (CRs) or partial remissions (PRs) confined to patients with low tumor burden even at suprasaturating doses that fully blocked CD33 binding sites for several weeks.11,12 Limited data suggested better efficacy in acute promyelocytic leukemia (APL) in patients with minimal residual disease (MRD) or when given at exceedingly high doses in non-APL AML.13,14 However, the development of lintuzumab was terminated after randomized trials showed no survival improvement when it was added to mitoxantrone, etoposide, and cytarabine (MEC) in patients with relapsed/refractory AML or to low-dose cytarabine (LDAC) in older adults with untreated AML.15,16 Similarly, the VEGF-A antibody bevacizumab did not improve response rates or survival in a randomized trial when added to standard induction chemotherapy in older adults with AML.17 Also unsuccessful were antibodies directed at CD123 (CSL360),18 FLT3 [IMC-EB10 (LY3012218)],19,20 or the oncofetal antigen EphA3 (KB004),21 which lacked antileukemic effects in most patients in phase 1 studies. Conversely, the CXCR4 antibody ulocuplumab combined with MEC yielded responses that compared favorably with historic controls treated with MEC alone among 43 patients with relapsed/refractory AML. Of note, 4 patients achieved a CR or CR with incomplete blood count recovery (CRi) after a single dose of ulocuplumab alone, demonstrating direct cytotoxic activity in a subset of patients.22 Some evidence of clinical antileukemia efficacy was likewise obtained with the KIR antibody IPH2101: at higher doses, this agent was associated with improved survival among 23 older adults with AML in first CR.23 A recombinant form of the same KIR antibody (IPH2102/BMS-986015, lirilumab) is being tested in ongoing clinical trials.
Clinical results with conjugated antibodies
Chemical modifications of monoclonal antibodies have long been pursued as a strategy to improve their clinical utility.2 Most widely tested are combinations with cytotoxic drugs (antibody–drug conjugates), toxic proteins from microorganisms or plants (immunotoxins), and radionuclides (radioimmunoconjugates).
Antibody–drug conjugates
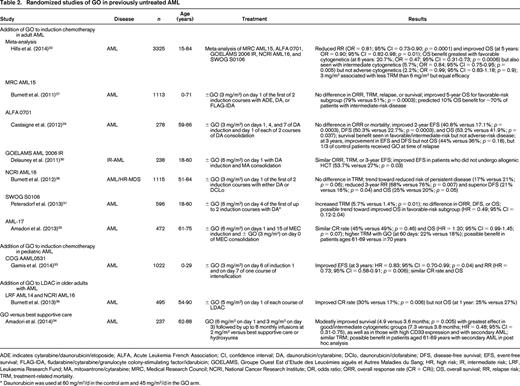
The most experience with any antibody-based therapeutic has been with GO, a humanized CD33 antibody conjugated to a calicheamicin-γ1 derivative via a hydrolysable linker. GO was initially granted accelerated US marketing approval in 2000 for adults aged >60 years with relapsed CD33+ AML who were not candidates for cytotoxic chemotherapy. Approval was based on an “overall” response rate [CR/CR with incomplete platelet recovery (CRp)] of 30% in 142 such patients entering phase 2 trials.24 Subsequent studies have confirmed single-agent activity of GO in newly diagnosed and relapsed/refractory non-APL AML, but overall response rates have usually not exceeded 25% to 35% and were occasionally very low.25 GO has higher activity in APL,26 likely because of the typically bright, uniform expression of CD33 on APL blasts, the lack of significant drug transporter activity, and possibly the expression of CD33 on underlying LSCs. Although many of the studies with GO were small and lacked comparative groups, several randomized trials have provided controlled data on the role of GO alone or in combination with other cytotoxic agents in various disease situations (Table 2). Most information comes from 5 studies that investigated GO in addition to intensive chemotherapy in adults with newly diagnosed AML.27-31 The 5 studies used GO in different schedules [3 mg/m2 on day 1 in 2 trials, 6 mg/m2 on day 4 in 2 trials, and 3 mg/m2 (up to a maximum of 5 mg/dose) on days 1, 4, and 7 in one trial]. An individual data meta-analysis of all 3,325 patients on these trials showed that GO significantly reduced relapse risk and improved survival. The reduction in relapse risk appeared greater with the fractionated dosing of GO than the nonfractionated schedule. Survival benefit was greatest in patients with favorable cytogenetics and was also seen in those with intermediate but not adverse cytogenetics. There was no increased 30-day mortality with the 3 mg/m2 dosing schedules, but an increase in early mortality was observed with the 6 mg/m2 dose in the Southwest Oncology Group (SWOG) S0106 trial.32 These findings are complemented by data from a large randomized pediatric trial [Children's Oncology Group (COG) AAML0531] in which the addition of GO to conventional chemotherapy significantly improved event-free survival and relapse risk but not remission rates or overall survival. In contrast to the adult trials, GO was beneficial in all risk groups in AAML0531.33 GO alone also provided a very modest benefit over best supportive care and hydroxyurea in untreated older adults considered “unfit” for intensive chemotherapy.34 Conversely, GO offered no benefit when used sequentially with MEC over MEC alone for older adults with untreated AML and was too toxic for those older than 70 years.35 It also did not improve survival when added to LDAC in this patient population despite nearly doubling the remission rate.36 Moreover, several studies found no benefit of GO during postremission therapy or as maintenance treatment.27,31,37-39 Because of the lack of overall improvement in outcome and concern over increased early mortality in the SWOG S0106 trial, GO was withdrawn from the commercial market in most countries in 2010. Discrepancies between S0106 and the other randomized trials in adult AML have been a topic of discussions in recent years, particularly with regard to the unexpectedly low early death rate in the control arm and the unequal doses of daunorubicin used in the two treatment arms of S0106. Nevertheless, despite repeated calls by many leukemia experts, GO has not (yet) been reintroduced.
The effectiveness of GO is curtailed by non-uniform drug conjugation, extrusion of the calicheamicin moiety via drug transporters, the limited amount of cell-surface CD33, and the relatively slow internalization kinetics of CD33/antibody complexes.25 Advancements in conjugation and linker technology and potency of the cytotoxic drug have resulted in SGN-CD33A, a humanized CD33 antibody with engineered cysteines carrying a synthetic DNA crosslinking pyrrolobenzodiazepine dimer via a protease-cleavable linker. Unlike GO, SGN-CD33A maintains activity in the presence of drug transporters in preclinical AML models.40 An ongoing phase 1 study primarily in older adults who either had relapsed/refractory AML or declined intensive therapy for newly diagnosed disease indicates that SGN-CD33A indeed has antileukemic activity: of 17 patients treated at a higher dose, this drug resulted in bone marrow blast clearance in 8 (47%), with 5 (29%) achieving either a CR or CRi.41 In contrast, phase 1 studies found low activity of AVE9633 (huMy9-6-DM4), a humanized CD33 antibody conjugated to a thiol-containing maytansinoid derivative, in adults with relapsed or refractory AML, and its clinical development has been terminated.42
Immunotoxins
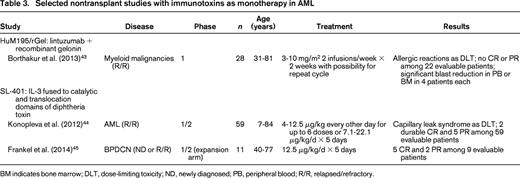
Under optimal experimental conditions, very few molecules of the toxic proteins used in immunotoxins are necessary to kill a eukaryotic cell, but such therapeutics have had limited efficacy in AML (Table 3). For example, HuM195/rGel, a CD33 antibody carrying recombinant gelonin, did not produce any CR or PR among 22 evaluable patients with advanced myeloid malignancies.43 Likewise, SL-401, a CD123-directed immunotoxin consisting of a fusion protein between IL-3 and the catalytic and translocation domains of diphtheria toxin, resulted in only 2 durable CRs and 5 PRs in 59 unselected patients with relapsed/refractory AML.44 Whether this drug is more efficacious in these patients when only minimal amounts of AML are present is currently unknown. More encouraging were results obtained in an expansion arm of that study in adults with blastic plasmacytoid dendritic cell neoplasm (BPDCN), a myeloid malignancy characterized by CD123 overexpression: among 9 evaluable patients, a single course of SL-401 yielded 5 CRs and 2 PRs, with responses lasting for a median of 5 months.45 Although additional studies are needed to elucidate why many immunotoxins are essentially ineffective in leukemia, postulated resistance mechanisms include neutralizing antibodies leading to clearance of circulating drug, limited antigen binding attributable to low-level target antigen expression, inability of the drug to infiltrate the bone marrow niche, and interference with any of the steps required for the toxins to reach the cytosol and exert their effects,46 including lysosomal degradation, which, based on preclinical models, may play a role particularly in multidrug-resistant leukemia cells.47
Radioimmunoconjugates
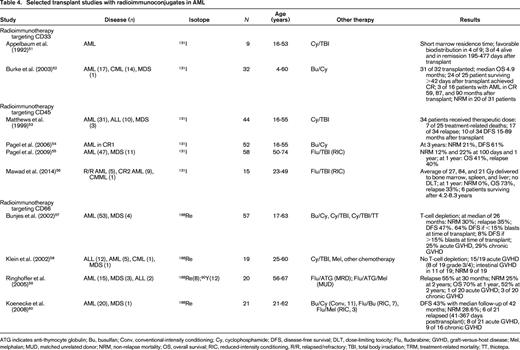
AML is exquisitely radiosensitive. Therefore, radionuclides are a natural choice to arm antibodies. Still, partly because of logistic hurdles and concerns over long-term safety, radioimmunoconjugates are currently not accepted widely as treatment for AML.48-50 The relatively long range of β-emitting radioisotopes (eg, 131I, 90Y, or 188Re) of up to 10 mm provides a “crossfire” effect on nearby cancer cells that do not express the target antigen or are not easily accessible to antibody. The much shorter emission range over just a few cell diameters and higher linear energy transfer of α-emitting radioisotopes (eg, 213Bi, 225Ac, or 211At) allows a more selective leukemia cell kill, but their availability is more limited and their radiolabeling chemistry more challenging. Several trials using radiolabeled antibodies to intensify conditioning before hematopoietic cell transplantation (HCT) have demonstrated tolerable toxicities and antileukemic activity, leading to their incorporation into transplant conditioning regimens even in older adults (Table 4).51-60 Although remissions have been achieved in most patients, the lack of well-controlled studies makes it difficult to know whether inclusion of radiolabeled antibodies indeed improves outcomes of HCT.8,61 More recently, “pretargeted” strategies that separate the targeting vehicle from the subsequently administered radioisotope62,63 (shown in preclinical models to improve tumor/organ ratios of absorbed radioactivity) are clinically investigated as a means to optimize delivery of radiation during transplant conditioning.64 In parallel, limited efforts have been undertaken to use α-emitting antibodies outside the HCT setting (Table 5).65 213Bi-labeled lintuzumab showed moderate antileukemic activity as a single agent but produced some remissions in a subsequent phase 1/2 study when used sequentially with cytarabine.66,67 Likewise, 225Ac-labeled lintuzumab, which can overcome some of the logistic challenges of the short half-life of 213Bi of only 46 min, demonstrated some antileukemic effects as a single agent and is being combined with LDAC in untreated AML.68,69
Evolving antigen-specific immunotherapies for AML
The randomized studies with GO validate CD33 as a target for antigen-directed immunotherapies at least for a subset of patients with AML and highlight the potential value of such approaches in this disease. Advances in structural engineering and chemical modification of antibodies give hope that improved antibody-based therapeutics will soon be available that could overcome the limitations of previous generation constructs. Adoptive immunotherapies using genetically modified immune effector cells and vaccination strategies complement the spectrum of evolving antigen-specific immunotherapies for AML. These efforts, summarized in the following sections, are paralleled by efforts to uncover new target antigens.70
New unconjugated and conjugated monospecific antibodies
For unconjugated antibodies, current efforts are focused on genetic manipulation to increase the interaction with activating Fc receptors, particularly CD16, and to improve antibody-dependent cytotoxicity mediated, for example, by NK cells.71 Fc-engineered antibodies targeting a variety of AML antigens (eg, CD33, CD38, CD96, CD123, CD133, CD135, CD157, CD300F, and T-cell immunoglobulin mucin receptor 3 [TIM-3]) have been preclinically tested recently, and some constructs targeting CD33 (MAb 33.1, BI 836858), CD123 (CSL362), and CD157 (MEN1112) have entered clinical testing.70,72 Instead of targeting AML antigens to elicit direct cytotoxic effects, some antibodies aim to amplify native immune responses, for example via modulation of programmed cell death-1 (PD-1)/programmed death ligand-1 (PD-L1) signaling, which may serve as a negative regulatory mechanism in AML. This possibility has been raised by preclinical data demonstrating that antibodies that block PD-L1 decrease AML burden in experimental mice.73 The PD-1 antibody CT-011 has already been assessed for safety in patients with relapsed/refractory hematologic malignancies including AML,74 and phase 2 trials of the PD-1 antibody nivolumab as monotherapy or CT-011 combined with a cell-based vaccine in patients with AML in remission have been initiated.
For drug-carrying antibodies, interest remains centered on CD33, with IMGN779, a CD33 antibody-drug conjugate that uses a DNA crosslinking indolino-benzodiazepine dimer, being another potential successor to GO. IMGN779 has shown high activity in preclinical AML models, particularly those with FLT3/internal tandem duplication (ITD) mutations,75 and may soon reach the clinic. In contrast, immunotoxins under preclinical development primarily aim at CD123 but also target other antigens (eg, CD64) and explore human toxins (eg, granzymes) to minimize neutralizing antibody formation, but it is unclear whether or when such agents will be introduced in the clinic.
Bispecific antibodies
One long-pursued engineering approach to increase antitumor activity is to create antibodies with dual affinities for a tumor cell antigen and an antigen on immune effector cells, such as cytotoxic T cells,2 directly engaging the latter in the elimination of the former. Initial success was limited by suboptimal effector cell recruitment and challenges with large-scale antibody production.76,77 These shortcomings may be overcome using small molecules consisting of single-chain variable fragments (scFVs), for example, bivalent or tetravalent antibodies that combine the scFVs on one [eg, bispecific T-cell–engaging (BiTE) antibodies] or two [eg, dual-affinity retargeting (DART) antibodies or tandem diabodies] polypeptide chains. These molecules bring polyclonal immune effector cells close to tumor cells and force formation of an immunological lytic synapse that triggers immune cell activation and proliferation and consequent destruction of attached tumor cells at a low effector/target cell ratio.77,78 High efficacy for some patients with acute leukemias was demonstrated in several positive trials with the CD19/CD3 BiTE antibody blinatumomab in chemotherapy-resistant Philadelphia chromosome-negative B lymphoblastic leukemia, which led to recent regulatory approval for this patient population. Efforts in AML primarily focus on CD3-directed agents that target CD33 or CD123, but a variety of alternate targets on tumor cells (eg, CLL-1 and FLT3) as well as immune effector cells (eg, CD16 or NKG2D) are being investigated. Preclinical studies with CD33/CD3-directed molecules, including the BiTE antibody AMG 330, have shown potent lysis of CD33+ AML cells together with healthy donor T cells or autologous T cells from AML patients and broad activity in human AML specimens.6,79-83 A CD123/CD3 DART molecule (MGD006) has entered phase 1 testing, and others, including AMG 330, will follow soon.
Adoptive immunotherapy
Adoptive immunotherapy using T cells expressing chimeric antigen receptors (CARs) or modified T-cell receptor (TCR) genes has drawn major attention in acute leukemias. CARs are hybrid single-chain receptor constructs containing an extracellular tumor antigen-recognizing domain linked to an intracellular component comprising the CD3 zeta chain with or without additional costimulatory endodomains to activate the immune effector cell during tumor cell binding.84 CD19-directed CAR T cells have shown very high remission rates and durable remissions in children and adults with B lymphoblastic leukemia, even among those who previously underwent allogenic HCT, demonstrating the potential of this approach.85,86 In AML, clinical experience is thus far very limited, with a study on 4 adults with relapsed disease treated with cytotoxic drugs followed by CAR T cells directed at Lewis-Y, a carbohydrate antigen overexpressed by malignant myeloid cells: of the 3 patients who had MRD at the time of cell infusion, one had a cytogenetic remission lasting 5 months, whereas the others had stable disease lasting 1.5 and 23 months, but ultimately all experienced disease progression despite persistence of CAR T cells and target antigen expression on myeloblasts.87 Although alternative antigens are explored,88,89 current efforts focus primarily on CD33 and CD123.90-95 Clinical trials testing CAR T cells directed at each of these antigens are ongoing, with one early report available of a patient who had a transient decrease in marrow blasts after receiving CD33-directed CAR T cells.96 Less explored are T cells expressing modified TCR genes. Besides leading to more physiological cellular activation, TCR-modified cells offer the advantage of being able to recognize intracellular epitopes expressed on the cell surface in the context of a peptide/major histocompatibility complex. This strategy is being studied in a phase 1/2 study in which patients with high-risk AML receive donor-derived Ebstein Barr virus- or cytomegalovirus-specific CD8+ T cells transduced with a high-affinity TCR specific for an HLA A*02:01-restricted WT1 peptide together with IL-2 after allogenic HCT. Early results on 9 treated patients, some of whom had measurable disease at the time of T-cell administration, suggest the possibility of antileukemic activity.97
Vaccines
Antibodies or immune effector cells directed against leukemia cell antigens are present in many AML patients.4 In some cases, these adaptive immune responses may have antileukemic effects.98 Although such observations support the potential value of active immunotherapy as treatment strategy in AML, vaccine-based therapies are still in their infancy. Most vaccination strategies target WT1, proteinase-1 (a specific PRTN3-derived peptide), and hyaluronan-mediated motility receptor (CD168) peptides. Case series and early-phase trials demonstrate the safety of such vaccines in AML patients with active disease, in remission after chemotherapy, and even after allogenic HCT and document immunologic and possible clinical responses in some cases.99-104 By introducing full-length antigens, cell-based vaccines may circumvent some of the limitations of peptide vaccines; still, early trials in which dendritic cells (DCs) were pulsed with leukemic lysates or directly derived from leukemic blasts yielded underwhelming responses.105-107 More effective may be autologous DCs transfected with WT1 mRNA, which yielded clinical responses, including remissions in patients who achieved a partial response with chemotherapy and molecular remissions in patients with residual disease. DC/AML fusion cell vaccines and “off-the-shelf” DC vaccines that are engineered from a myeloid leukemic cell line endogenously expressing a range of AML-associated antigens have also been used in early-phase trials,108-111 but additional studies are needed to demonstrate unequivocally that improved disease control can be achieved with these agents.
Open questions
With the many antigen-specific immunotherapeutics currently under development, we can be hopeful that we can soon expand the armamentarium for treatment of AML. Nonetheless, a number of important questions remain open at this time and need to be addressed in future investigations. Perhaps foremost is the question of the ideal target antigen(s). It is likely that there is no single optimal antigen in a disease as heterogeneous as AML; the clinical experience with GO is an important reminder that AML cannot be regarded as one disease. For any particular antigen, only a subset of patients may be suitable, and the choice of target antigen may depend on the purpose of the therapy, that is to say whether the primary intent is to reduce the burden of leukemia cells in a patient with active disease or to eradicate MRD and the very rare underlying LSCs. Elimination of LSCs may be an ideal goal for immunotherapies but will require a deepened understanding of the immunophenotypic and functional properties of these elusive cells. LSC-directed therapy may be most successful if the immunophenotypic properties of LSC are clearly distinguishable from normal hematopoietic stem cells; the particular susceptibility of APL to CD33-targeted immunotherapies may support this notion.7 If it is necessary to target more than one antigen for maximum treatment benefit, in what sequence should the therapeutics be used, and how can they be combined with conventional chemotherapeutic or immune-stimulating drugs to achieve optimal effects? Because the display of target antigens may change on AML cells and normal cells during treatment, for example, as the cell cycle status changes, pretreatment antigenic profiles may not adequately reflect expression patterns during therapy. Of course, the exact antigenic profile of some of the targeted cells such as LSCs may not be known at all given their rarity and our current lack of ability to isolate/enrich them effectively. Especially in light of the diversity of explored therapies, another open question is which class of therapeutics will provide optimum outcomes, augmenting the anti-AML effect while minimizing toxicity to normal cells. Life-threatening toxicities with some passive immunotherapies, particularly gene-modified immune effector cells, are now well recognized. Very likely, the ideal format of immunotherapy will depend on many factors, including target antigen, disease stage, and patient characteristics. Finally, if AML-associated antigens such as CD33 or CD123 are targeted, what degree of supportive care is needed? Highly potent therapeutics, such as bispecific antibodies and CAR T cells, will have profound effects on normal hematopoietic cells expressing these antigens and cause prolonged cytopenias. This will put high demands on supportive care measures in a vulnerable patient population. Some therapeutics may even require autologous or allogenic stem cell rescue for safe administration to abrogate their effects on normal hematopoietic cells and/or immune effector cells and therefore could primarily play a role during transplant conditioning. Answers to such questions will ultimately be required to optimize the clinical use of antigen-specific immunotherapies in AML and to ensure that they become an effective tool for this disease for which outcomes have remained unsatisfactory.
Acknowledgments
We thank Drs. Irwin D. Bernstein, Elihu H. Estey, and Wendy Stock for critical reading of this manuscript.
R.B.W. is a Leukemia & Lymphoma Society Scholar in Clinical Research.
Correspondence
Roland B. Walter, Clinical Research Division, Fred Hutchinson Cancer Research Center; 1100 Fairview Ave N, D2-190, Seattle, WA 98109-1024. Phone: 206-667-3599; Fax: 206-667-6519; e-mail: rwalter@fredhutch.org.
References
Competing Interests
Conflict-of-interest disclosures: S.A.B. reports no conflict of interest. R.B.W. has received research funding from Amgen, Amphivena Therapeutics, and Seattle Genetics and has consulted for Covagen, Amphivena Therapeutics, Pfizer, and AstraZeneca.
Author notes
Off-label drug use: Alemtuzumab, bevacizumab, and GO for the treatment of AML.