Abstract
Recent approvals of several oral targeted agents have revolutionized chronic lymphocytic leukemia (CLL) therapy. However, CLL patients continue to progress; particularly, 4% to 20% of previously treated CLL patients undergo transformation into high-grade lymphoma. Richter transformation is defined as a transformation of CLL into aggressive lymphoma, most commonly diffuse large B-cell lymphoma. These patients typically have poor response to traditional chemotherapy used to treat de novo diffuse large B-cell lymphoma and similar or shorter overall survival (median 3-11 months) in the era of novel agents. Here, I review the contemporary literature on Richter transformation, particularly in the context of novel agents used in CLL, and discuss the management approach for these patients.
Learning Objectives
Understand the incidence, diagnosis and pathobiology of Richter transformation in patients receiving novel agents for CLL
Introduce a new algorithm for the management of Richter transformation based on available molecular and therapeutic data
Introduction
Since the initial description by Maurice Richter in his article in the American Journal of Pathology in 1928,1 chronic lymphocytic leukemia (CLL) transformation into a more aggressive lymphoma has been recognized as Richter syndrome (RS). The 2008 World Health Organization classification of hematopoietic tumors defines RS as the transformation of CLL into a more aggressive lymphoma. This definition remains the same in the 2016 revision of the World Health Organization classification of lymphoid neoplasms.2 The majority of RS represents a transformation from CLL into diffuse large B-cell lymphoma (DLBCL), although Hodgkin lymphoma (HL), plasmablastic lymphoma, or other rare lymphomas have been reported. Here, I will focus on reviewing the published data on the biology and therapy for DLBCL transformation from CLL, briefly Richter transformation (RT), prior to and now in the era of novel agents. I will also briefly discuss HL transformation.
Incidence of RT has not decreased in the era of novel agents
Historically, in the era prior to the use of novel oral agents, including the Bruton tyrosine kinase (BTK) inhibitor ibrutinib (Ibr), the phosphoinositide 3-kinase (PI3K) inhibitor idelalisib, and the B-cell lymphoma-2 (BCL-2) inhibitor venetoclax (Ven), studies that tested the prevalence of RT mainly focused on CLL patients who had received prior chemotherapy or chemoimmunotherapy (CIT). With a median follow-up of 3 to 12 years, the prevalence of RT varied from 1% to 10.7%. For example, after a median observation time of 69 months, RT developed in ∼5% of 1450 CLL patients treated within the context of several German CLL trials (CLL4, CLL5, CLL8, and CLL2m) in which patients received first-line chemotherapy.3 With a longer median follow-up of 12 years, the occurrence of RT was 8% in CLL patients treated with first-line fludarabine, cyclophosphamide, and rituximab at MD Anderson Cancer Center (MDACC).4 Using a different approach, the Mayo Clinic CLL Group investigated the prevalence of RT in a cohort of 1641 newly diagnosed CLL patients with a median follow-up of 4 years. A total of 2.3% of newly diagnosed CLL patients developed RT, with a median time to development of 1.8 years. The incidence of RS was 0.5% per year in untreated patients (approximately half of this RT cohort) and 1% per year in treated patients.5,6
In the era of novel targeted agents, the reported incidence of RT varies (3% to 20%) among clinical trials or retrospective studies. In the initial studies from The Ohio State University (OSU), 6.5% of 308 CLL patients receiving Ibr developed RT within 18 months.7 Subsequent follow-up data reported that 6% of patients (n = 132) developed RT after a 3-year follow-up; the majority of RT cases (7% in previously treated CLL, 3% in therapy-naive CLL) were reported in the relapsed or refractory cohort and occurred within the first year of Ibr initiation.8 Similarly, ∼5% of patients treated with Ibr developed RT within 15 months of therapy in several studies, with a median follow-up between 2 and 5 years.9-11 Importantly, Jain et al12 reported an incidence of RT of ∼3% in therapy-naive CLL patients treated with Ibr (n = 68) at MDACC after a 3-year follow-up. In contrast to the 3- to 5-year follow-up trial of Ibr, data published for CLL transformation after treatment with the BCL-2 inhibitor Ven had a shorter follow-up (∼2 years). A total of 23% of patients developed RT within the first year of Ven therapy in a combined cohort of 67 relapsed or refractory CLL patients.13 Subsequent study testing the efficacy of Ven in del(17p) CLL reported ∼10% RT occurrence.14 When the combination of Ven and rituximab was evaluated, 3% or 10% of CLL patients developed RT in 2 studies.15,16 In order to eliminate preexisting RT prior to trial enrollment, 2-deoxy-2-[18F]fluoroglucose (FDG) positron emission tomography/computed tomography (PET/CT) scan and subsequent biopsy were performed to exclude potential RT in a trial testing Ven in progressive CLL that received prior Ibr or idelalisib. Approximately 5% of enrolled patients developed transformation while receiving Ven.17
In summary, the reported incidence (3% to 20%) of RT in the era of novel agents does not appear to be decreased in comparison with the incidence of RT in the CIT era. Typically, RT occurs early while receiving novel targeted agents with a caveat of possible enrollment of preexisting RT in early trials. The incidence of RT appears to be lower in patients who received a first-line kinase inhibitor than in relapsed/refractory CLL patients. Whether novel oral targeted therapies alter the incidence of RT remains unclear. This question will hopefully be answered after revealing the follow-up data of several phase 3 trials to compare CIT with novel agents (North America Alliance A041202 and E1912, United Kingdom FLAIR, and the German CLL13 trial).
Diagnosis of RT requires tissue biopsy and high clinical vigilance
When do I suspect RT in CLL? The clinical presentation of RT varies from worsening discordant lymphadenopathy or new-onset cytopenia to rapid clinical deterioration (constitutional symptoms, rising lactate dehydrogenase levels, and hypercalcemia). A comprehensive evaluation with PET/CT scan and bone marrow biopsy is recommended for evaluation of potential transformation. In the era of novel agents, a marked increase in lymphadenopathies, particularly within the first or second year of therapy, should prompt evaluation for possible RT. An image-directed biopsy is essential to establish the diagnosis. It is also recommended to obtain CLL fluorescence in situ hybridization (FISH), TP53 somatic mutation status, and clonality testing between RT and CLL at the time of evaluation, because these results may provide additional prognostic indications.
The value of PET/CT to aid in RT diagnosis has been demonstrated in several studies in the CIT era. Specifically, a median standardized uptake value (SUV) was found to be 3.5 or 14 for CLL or DLBCL, respectively.18 A SUV ≥5 was associated with inferior outcomes in CLL. However, a SUV of 5 for index lesions detected on PET/CT was associated with a high (97%) negative predictive value but low positive predictive value (53%) for RT diagnosis,19 indicating the necessity of tissue biopsy for a PET-avid lesion. A subsequent study showed that an SUV of 10 on PET scan can distinguish RT from CLL with 91% sensitivity and 95% specificity.20 Falchi et al21 found that a maximum SUV (SUVmax) ≥10 was associated with poor survival regardless of whether the diagnosis was RT or aggressive CLL.
In the era of novel agents, a SUVmax of 10 as radiographic criteria for potential diagnosis of RT has been challenged. In a recent investigation testing the predictive value of PET in CLL patients who progressed after BTK or PI3K inhibitor therapy, SUVmax ≥10 was associated with 26% sensitivity and 82% of specificity for an RT diagnosis.22 Specifically, the RT DLBCL variant was confirmed in 5 out of 8 patients with an SUVmax of 5 to 9 and in 3 out of 8 patients with an SUVmax >10. These data emphasize that a specific SUV alone cannot be used to rule out (or rule in) RT and indicate the diagnostic necessity to biopsy predominant index lesions. I recommend choosing the index lesion with the highest SUV uptake first. If an FDG-high lesion (SUV >10) is not present, then an index lesion with intermediate to high PET FDG avidity (SUV 4-10) should be biopsied if clinical suspicion is high. The optimal way to biopsy the index lesion on PET scan depends on the location of the lesion. Wherever possible, excisional biopsy is always preferred. Samples obtained with fine-needle aspiration commonly are not diagnostic, because they lack complete tissue structure. Partial sampling of regions containing discrete large cells in an expanded proliferation center, typically present in aggressive CLL, may lead to a false-positive diagnosis of RT. Similarly, partial sampling may miss the foci of true transformation and lead to a false-negative diagnosis of RT. For patients whose only sites of FDG-avid disease are not easily accessible for an excisional biopsy (eg, retroperitoneal or abdominal), a CT- or ultrasound-guided large-bore core needle biopsy should be pursued.
Pathologically, the diagnosis of RT can be challenging and needs to be reviewed by an experienced hematopathologist. The current World Health Organization Classification of hematopoietic and lymphoid tissues does not provide strict criteria to diagnose RT. Because progressive CLL can be associated with an increased percentage of discrete large cells, these large cells could represent immunoblasts in enlarging proliferation centers in the background of CLL. The diagnosis of RT should be restricted to cases with confluent sheath of central blast– or immunoblast-like large neoplastic lymphoma cells. These diagnostic challenges were illustrated in several studies, in which central pathological review confirmed RT in only 80% of cases previously identified as RT.23,24 Given these findings, the criteria to diagnose RT were proposed based on the diagnostic criteria for DLBCL and include (1) large B cells with a nuclear size equal to or larger than macrophage nuclei or more than twice the size of a normal lymphocyte and (2) a diffuse growth pattern of large cells.
In summary, early progression with increased lymphadenopathy in CLL patients receiving a novel agent should sound the alarm for possible RT. Biopsy of an index lesion identified on PET scan is required to confirm the diagnosis. Pathologically, the diagnosis of RT requires careful review by an experienced hematopathologist.
RT development in the era of novel agents shows predominant TP53 disruption and possible involvement of B-cell receptor (BCR) signal with expansion of BTK mutant clones
The molecular mechanisms underlying RT development from CLL are not completely understood. Transformation is likely a multistep process driven by clonal evolution of CLL disease and additional genetic events that either spontaneously occurred or were triggered by prior therapies. Alternatively, RT can arise from a new B-cell clone unrelated to CLL clonal B cells. The majority (≥80%) of RT with DLBCL subtype is clonally related. Molecular analyses show RT follows a linear evolution in more than half of patients and less for a branching evolution.25,26
Based on accumulated data published by Rossi, Chigrinova, and Fabbri et al in the CIT era,25-27 molecular biomarkers associated with the RT DLBCL variant are distinct from de novo DLBCL. Disruption of the TP53 gene by either deletion or mutations is detected in up to 60% of RT cases vs 10% to 20% in de novo DLBCL. TP53 disruption is more commonly found in clonally related RT. Gain-of-function NOTCH1 mutations are reported in ∼30% of RT, frequently among patients with trisomy 12. CDKN2A/B deletion, found in ∼30% of RT cases, can coexist with TP53 disruption or NOTCH1 mutation. Overexpression of BCL-2 is frequently detected26 ; however, amplification or gene translocation involving BCL-2 is not common as reported in the germinal center B-cell subtype of DLBCL. MYC is frequently aberrant in 50% of DLBCL RT caused by gene rearrangement t(8:14), structural alteration (∼30%),25-27 or deletion of the MYC negative regulator MGA (∼10%).25,28,29 In addition, Notch activation either by mutation or increased expression implicates MYC signal activation.30 These molecular abnormalities imply the role of apoptosis regulation, DNA repair, and cell-cycle regulation in the development of RT. Biased usage of stereotyped immunoglobulin genes in subset 8 (IGHV4-39/IGHD6-13/IGHJ5) is present in a subset of RT,31,32 suggesting that BCR signaling contributes to the development of RT. Subset 8 patients have enriched trisomy 12 and NOTCH1 mutation, which represent the second most common CLL FISH finding (after del(17p)) associated with RT development.32 In addition, the majority of DLBCL RT patients have unmutated immunoglobulin heavy-chain variable region gene (IGHV) and are negative for Epstein-Barr virus (EBV) staining by in situ hybridization of EBV-encoded RNA.
Scarce data are available for RT that develops with novel agents. Combined data showed that >70% post-Ibr RT DLBCL patients had TP53 disruption7,9,11,12,33,34 (including 8 out of 14 RT cases from OSU, 6 out of 6 RT cases from the University of Chicago, 4 out of 9 RT cases from MDACC, 4 out of 5 RT cases from the Mayo Clinic and 5 out of 6 RT cases from the National Institutes of Health). All RT patients in the OSU cohort had complex karyotypes, and 5 out of 40 RT patients (∼12%) had trisomy 12 among the combined cohorts. Complex karyotypes and the presence of near tetraploidy are associated with RT development in Ibr-treated patients.35 All 8 RT patients who were tested were clonally related to CLL. Davids et al36 presented the largest cohort of RT patients (∼71 cases) in the era of novel agents, including 59 post-BTK inhibitor cases and 6 post BCl-2 inhibitor or PI3K inhibitor cases each. Approximately 50% of RT cases have del(17p), 75% of cases have complex karyotype, and 25% of cases have trisomy 12. Among the limited RT patients tested, a TP53, NOTCH1, or SF3B1 mutation was detected in 80% of cases in each category. A spread of IGHV family distribution was detected without particular enrichment of one specific IGHV in this RT cohort.
Even though BTK and PLCG2 mutations are frequently uncovered in CLL resistant to Ibr,37 an understanding of the contribution of these mutations along the BCR signal pathway to RT development has just begun to emerge. Kadri et al33 first reported the genomic abnormalities of 6 patients from the paired CLL blood and RT-involved tumor tissue after progression on Ibr. The majority of genetic aberrations (60% to 95%) were found in both RT tissue and CLL leukemia cells. An additional 1 to 15 RT-specific mutations were identified in RT tissues only.33 8q gain (MYC) is the only recurrent RT-specific aberration. In 4 out of 6 RT cases, del(18p) was detected in CLL leukemic cells before Ibr treatment. Among patients with RT who carried BTK mutations (n = 4) in their CLL cells, the paired RT tissues also carried the same BTK mutations in 2 patients. In the third patient, the RT tissue had a major clone of BTKC481Y, whereas a minor clone of BTKC481S was identified in blood CLL cells. In addition, all BTK mutations uncovered in this cohort had coexisting TP53 disruption. Taken together, these data indicate that expanding clones carrying BTK mutations contribute to RT development in a major fraction of post-Ibr patients, and TP53 disruption may provide a permissive environment for the outgrowth of these clones. Similarly, among 14 RT post-Ven patients,13 10 (71%) had TP53 disruption, and 5 out of 8 (63%) had complex karyotypes. BCL-2 expression by immunohistochemistry was evident in most RT DLBCL cases. In another study that analyzed 8 progressive patients receiving Ven,38 4 were RT DLBCL variant, and all 8 (100%) had evidence of TP53 disruption. In addition, all progressive patients, including those with RT, showed evidence of developing additional mutations and genomic instability. Loss of CDKN2A/B occurred in 5 out of 8 patients. Additional mutations in BRAF and CD274 amplification were detected.38
Despite advances in understanding the molecular aberrations in tumor biology, much remains to be investigated in the immune evolution of RT patients. Our preliminary data showed that PD-L1 expression is increased in RT-involved nodal tissue in comparison with CLL nodal tissue. Clonality of T-cell receptor repertoire decreased in the RT cohort in comparison with CLL, indicating that a diversification of T-cell receptor repertoire occurs during CLL transformation into RT,39 possibly secondary to newly formed tumor antigens. These results are consistent with the above finding that more tumor mutations are detected in RT.
In summary, RT in the era of novel agents primarily arises in the genetic background of TP53 disruption and complex karyotype. Similar to the data obtained prior to novel agents, MYC activation (8q gain or NOTCH1 mutation) and loss of CDKN2A/B likely play major roles in RT development. Of particular interest, BTK mutant expansion occurs in a significant portion of RT tissue after BTK inhibitors, indicating a contribution of BCR signal in RT. In addition to the above abnormalities, gaining additional mutations in individual RT patients facilitates RT development and possible immune evasion from host immunity.
Prognosis of RT in the era of novel agents is extremely poor
Clonally related RT patients (>80% of RT DLBCL) typically have a poor response to traditional chemotherapy used to treat de novo DLBCL and short survival (<1-2 years). However, clonally unrelated DLBCL RT patients have a similar response to rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) as de novo DLBCL patients and have much better survival (∼5 years).27 Thus, it is critical to determine the clonal relationship of RT with CLL to choose optimal therapy. However, a determination of the clonal relationship requires the analyses of immunoglobulin heavy-chain gene rearrangement in RT-involved tissue and preexisting or concurrent CLL samples that are rarely available. Alternative tests to determine potential clonal relationships have been proposed. He et al40 first tested PD-1 expression by immunohistochemistry in 39 CLL/small lymphocytic lymphoma (SLL) patients, 15 DLBCL RT patients, and 26 other DLBCL patients in a Mayo Clinic RT study. In CLL/SLL, neoplastic B-cell PD-1 expression was weak and restricted to paraimmunoblasts within proliferation centers. Increased expression of PD-1 was found in 12 out of 15 cases(80%) of DLBCL RT tumor cells. In contrast PD-1 expression was only seen in 1 out of 26 other de novo DLBCL. An excellent correlation (90% concordance) was observed between neoplastic B-cell PD-1 positivity and a molecularly defined CLL/SLL clonal relationship in DLBCL RT. The presence of classical immune markers for CLL (eg, CD5 and CD23) can suggest a clonal relationship between RT and CLL. However, these markers were found to be lost in a significant portion of DLBCL RT tissue samples regardless of clonal relationship with the underlying CLL.41
Of 86 patients with RT seen in the CIT era by Rossi et al,27 ∼60% to 70% were treated with CHOP or R-CHOP or second-line non-HL (NHL) chemotherapy. Using multivariate analysis, TP53 disruption was identified as the only molecular marker that has distinct prognostic impact regardless of clonal relationship, possibly secondary to known TP53-mediated chemorefractoriness in CLL.42 Eastern Cooperative Oncology Group performance status (ECOG PS) and complete response (CR) to induction therapy were the other 2 important prognostic factors. These data were further confirmed by Chigrinova et al25 in their analysis of 60 RT patients, where 75% were treated with CHOP-like or second-line NHL regimens. Among patients who had long-term follow-up, aberrations in TP53/CDKN2A predicted worse overall survival. The significance of TP53 disruption to predict worse overall survival of RT patients treated with R-CHOP,43 R-EPOCH (rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin),44 CHOP-O (CHOP with ofatumumab),45 or other chemoimmunotherapies46-48 had been repetitively confirmed. In addition, several studies revealed that the number of prior therapies is highly prognostic.44,45,49 Specifically, in a National Cancer Research Institute phase 2 study of CHOP-O,45 patients who were therapy naive had a significantly superior response rate and survival than patients who had prior therapies. In an MDACC retrospective analysis of their RT cohort, a prognostic score based on 5 adverse risk factors (performance status >1, increased lactate dehydrogenase, platelet count ≤100 × 109/L, tumor size >5 cm, and ≥2 prior therapies) are capable of stratification of RT patients into low-risk (0-1), intermediate-risk (2), and high-risk groups (3-5) to predict differences in survival time.49
The prognostic scores described above were validated in the CIT era, but not tested for their validity to predict survival in the era of novel agents. Limited available data showed most RT cases (over 90%) in the new era are clonally related, >70% RT cases have TP53 disruption, and almost all RT cases have complex karyotypes.
Multiple studies reported universal poor outcomes with a median survival of ∼4 months when RT developed while receiving Ibr.7,12,36 Complex karyotype and fludarabine refractoriness were identified to be key risk factors for RT development on Ven. A response rate of 40% was observed when treated with chemotherapy, and the median survival of these RT DLBCL patients post-Ven therapy was ∼11 months. Three patients who later started BTK inhibitor therapy, including 2 patients who had received a transplant, had relative long-term survival (3-4 years). These RT patients typically present bulky nodal or extranodal diseases after Ibr or Ven therapy, similarly to patients with highly aggressive B-cell lymphoma. Concurrent resistant CLL in bone marrow or blood are common. These clinical characteristics implicate the difficulty and complexity to manage these RT patients effectively.
Therapy options for RT developed in the era of novel agents for CLL
There have been no randomized trials investigating therapeutic agents for RT that developed from prior CLL. All published evidence was from single-arm prospective trials with a small number of patients or from retrospective studies. Among the published trials, most were from the CIT era prior to the use of novel agents. Historically, most RT DLBCL cases were treated with a combination of chemotherapies used to treat de novo DLBCL, such as R-CHOP or R-CHOP–like (eg, R-EPOCH) regimens. These data were thoroughly reviewed by multiple prior reviews.50-53 In summary, R-CHOP as a first-line therapy for RT showed a response rate of 50% to 60% and a median overall survival of 15 to 21 months.43 The substitute of rituximab with ofatumumab did not result in a higher response rate (46%) or longer survival (11 months).45 A retrospective study showed that front-line R-EPOCH therapy had a response rate of 39%, a progression-free survival (PFS) of 3.5 months, and overall survival of 5.9 months.44 The addition of a CLL-directed chemotherapy agent in the regimen oxaliplatin, fludarabine, cytarabine, and rituximab (OFAR) was associated with a response rate of 38% to 50% and a median survival of 6 to 8 months.46,48 Dose intensification with R-hyper-CVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, adriamycin, and dexamethasone)47 or including platinum in the regimen in DHAP (dexamethasone and cytarabine) or ESHAP (etoposide, solumedrol, cytarabine, and platinum)54 increased CR rate but was associated with severe hematological toxicity, increased infection, and relative high treatment mortality, resulting in a similar or shorter median survival.
Given the short duration of response achieved with chemotherapy, autologous and allogeneic stem cell transplantation has been proposed as postinduction strategy with the goal of maintain durable remission in DLBCL-type RT. A retrospective analysis by the European Group for Blood and Marrow Transplantation showed a subset of patients benefited from these procedures.55 The estimated 3-year survival rate is 36% after allogenic stem cell transplant (allo-SCT) and 59% after autologous stem cell transplant (auto-SCT). Importantly, retrospective analysis of transplant data are subject to selection bias, because transplant patients were selected based on good clinical performance status and at least partial response (PR) to prior chemotherapy. Unfortunately, the majority of RT patients (80% to 90%) will either not achieve a good enough response to be able to proceed with transplantation or their comorbidities or age will prevent them from undergoing transplantation. This was illustrated by results from MDACC; only 20 out of 148 biopsy-confirmed RT patients (14%) underwent SCT. Patients who underwent allo-SCT in CR or PR have good survival (75%) at 3 years.49
In total, trials performed prior to the use of novel agents showed that R-CHOP or R-CHOP-like regimens are still the standard therapy to treat RT. Fit and young patients who achieve CR or good PR potentially benefit from a postinduction strategy (allo-SCT or auto-SCT). However, these data have not been validated in the era of novel agents for CLL, in which most RT cases appear to be associated with poor prognostic markers. Several trials or retrospective studies have tested the role of novel agents in the era of novel agents for CLL.
Checkpoint inhibitors
Preclinical evidence support that exhausted T cells contribute to the immunodeficiency status of CLL. The Mayo Clinic CLL Group reported the first trial of a PD-1 blocking antibody, pembrolizumab, in CLL (n = 16) and RT (n = 9) patients.34 The overall response rate in patients with RT was ∼40%, whereas no responses were seen in CLL patients. In particular, all responses to pembrolizumab were observed in CLL patients who developed RT after progression on Ibr (4 out of 6). Five out of 9 patients had relapsed/refractory RT disease before the initiation of pembrolizumab. Overall survival for the whole RT cohort was ∼11 months since the trial enrollment. There is a trend of better survival for these RT patients post-Ibr therapy. Increased PD-L1 expression in the nodal tissue microenvironment of RT was detected in responders. PD-1 blockade appears to be capable of inducing nodal reduction in a portion of RT patients, but not associated with marrow CLL response. Thus, a combination of CLL therapy with checkpoint inhibitors is needed to effectively control both diseases. This was illustrated in a combination trial of another PD-1 antibody (nivolumab and Ibr) conducted by MDACC.56 Three out of 5 RT patients (60%) achieved a PR. Similarly, a combination trial of nivolumab and Ibr was tested in ∼20 RT patients in a subsequent trial.57 An overall response rate of 60% but relative short PFS (∼4 months) were observed. Here, a portion of RT patients who progressed on Ibr developed a clinical syndrome of fever and constitutional symptoms associated with stopping Ibr.58 These constitutional symptoms can be controlled by restarting or continuing Ibr despite apparent progression of disease on Ibr. In these patients, therapy with checkpoint inhibitor alone does not appear to be enough to control aggressive disease. A combination of alternative CLL therapy (eg, BCL-2 inhibitor) with Ibr or another BTK inhibitor and checkpoint inhibitor may be needed to control complex CLL and RT disease. Further clinical trials (www.clinicaltrials.cog #NCT02332980, #NCT02420912, #NCT02535286, #NCT02362035, and #NCT02846623) utilizing different combination approaches are ongoing to further test the efficacy of checkpoint inhibitors in RT.
Targeted inhibitors: BTK inhibitors, BCL-2 inhibitors, or nuclear export inhibitors
Given the refractory nature of the majority of RT patients to chemotherapy, BTK inhibitors have been evaluated in RT. Initial case reports from the Mayo Clinic CLL Group showed 3 out of 4 refractory RT patients responded to Ibr or a combination of Ibr with steroids. The median duration of therapy was 6 months.59 Another BTK inhibitor with more specific activity to target BTK, acalabrutinib, was tested in the ACE-CL-001 phase 1/2 trial. The overall response rate to acalabrutinib among 29 RT DLBCL patients was 38% and was associated with a PFS of 3 months, and the median duration of response was 5 months (#NCT02029443).60 A combination of Ibr with obinutuzumab with or without CHOP is being tested for RT (#NCT03145480). In the M12-175 phase 1 study, 7 DLBCL-type RT patients were treated with escalating doses of the BCL-2 inhibitor Ven, achieving a response rate of 43% with unknown duration.61 A combination trial of Ven with R-EPOCH for RT DLBCL (#NCT03054896) is ongoing. Alteration in protein transport between the nucleus and cytoplasm plays an important role in the regulation of tumor-suppressor proteins. Exportin 1 (XPO1) is the nuclear exporter of TP53, and XPO1 mutation has been identified in CLL patients. Selinexor is a selective inhibitor of nuclear export. In a phase 1 trial, selinexor showed activity in 40% of DLBCL RS patients (2 out of 5 patients had a PR) who were refractory to the previous chemotherapy regimen.62
CAR-T or bispecific antibody
Chimeric antigen receptor T (CAR-T) cells targeting CD19 have shown a response rate of 60% to 70% in CLL.63,64 In particular, CLL patients who progressed on Ibr appear to respond at a similar rate (74%), with a complete response rate 21%.65 In this trial, 5 RT patients were treated and 3 had a CR or PR. Thus, it represents a significant therapeutic advance in highly refractory CLL or RT patients. This approach is being further tested in the JCAR017 trial (#NCT03484702). Limited data regarding the use of different CAR-T therapies in the setting of RT has shown that among 2 RT patients treated with different CAR-T products, one had disease progression66 and the other presented disease evolution to plasmablastic lymphoma.67 Blinatumomab, a bispecific T-cell engager antibody designed to direct cytotoxic T cells toward CD19-expressing B cells, has been approved for acute lymphoblastic leukemia. This agent has also shown to induce 50% to 60% overall response in relapsed/refractory DLBCL.68 A RT case has been published that was treated effectively by blinatumomab and then followed by stem cell transplant.69 Further study is ongoing to test the efficacy of this agent in RT patients (#NCT03121534).
In summary, novel agents are currently being explored in RT. In particular, novel therapies to activate T cells, either in the format of checkpoint inhibitors or CAR-T/bispecific antibodies, appear to be promising. However, knowledge gaps are present in our understanding of toxicity, biomarkers to select patients who likely benefit, and how to best sequence the novel therapies with stem cell transplant. Further studies addressing these knowledge gaps are needed.
Suggested approach to manage RT with DLBCL subtype
Given the lack of data for DLBCL RT in the era of novel agents, it is not easy to recommend a standard approach to manage RT patients. Further study to gather enough knowledge of the tumor biology and immune microenvironment is needed to design more effective therapies. However, certain data can be extracted from the data obtained in the CIT era to aid in our current understanding of RT in the era of novel agents. First, TP53 disruption is present in a higher proportion (possibly in the context of complex karyotype) in RT patients, and continues to predict a poor response to standard chemotherapy. Second, the pathological diagnosis of RT in the era of novel agents remains same, requiring evaluation by a hematopathologist who has expertise to evaluate CLL and DLBCL pathology. In the era of novel agents, clinical suspicion is high for RT if a CLL patient develops enlarging lymphadenopathy in the first several years of therapy. A 18FDG PET/CT scan should be performed, and an excisional biopsy of an index lesion is required for diagnosis. Additional tests that can inform therapy selection include clonality, karyotype, TP53 mutation, CLL FISH, and BTK resistant mutation. These tests should be performed preferably on the RT-involved nodal tissue.
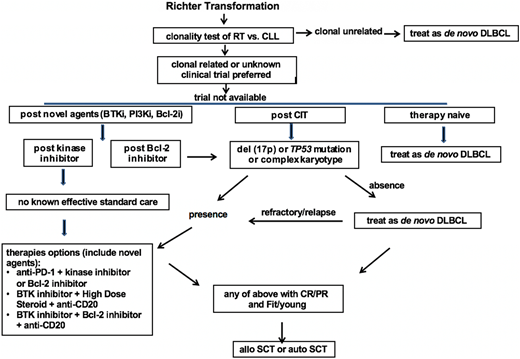
Based on the relative strong evidence gathered from the largest series of RT patients, chemotherapy used to treat de novo DLBCL, typically R-CHOP or R-CHOP–like regimens containing anthracycline or platinum, is recommended as the first-line therapy for clonally unrelated DLBCL RT. For clonally related RT or cases with unknown clonality, clinical trials are preferred if available. If there is no suitable trial available, then the recommendation is based on the status of their prior therapy and prognostic factors. In therapy-naive RT, standard therapy for de novo DLBCL is recommended as the first-line therapy. For RT that develops after prior CIT directed to CLL, such as fludarabine, cyclophosphamide, and rituximab/bendamustine and rituximab (BR)/pentostatin, cyclophosphamide, and rituximab (PCR)/fludarabine and rituximab (FR)/rituximab and chlorambucil (R-CLB), therapy is recommended based on the status of TP53 disruption and karyotype results. In the case of absence of TP53 disruption or complex karyotype, an R-CHOP–like regimen is recommended. In the case of the presence of TP53 disruption or complex karyotypes, novel combinations are recommended. These combinations can be a targeted inhibitor with an anti-PD-1 antibody, a BTK inhibitor with a BCL-2 inhibitor, or a BTK inhibitor with solumedrol and a monoclonal antibody directed to CD20. In RT that develops after therapy with a BTK inhibitor (eg, Ibr), I recommend the novel combinational approach listed above given the extremely poor outcomes observed using an R-CHOP–like regimen in these patients. In RT that develops after BCL-2 inhibitor therapy, I recommend the same approach listed above for post-CIT RT (Figure 1).
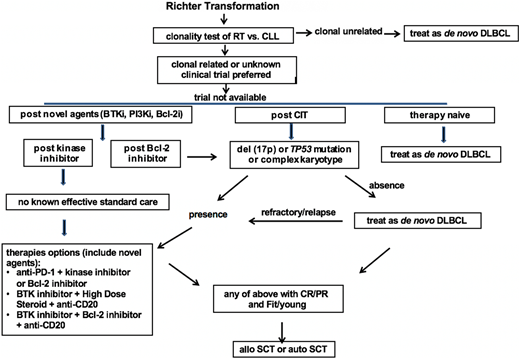
Proposed algorithm for the management of RT DLBCL in the era of novel agents of CLL. Bcl-2i, Bcl-2 inhibitor; BTKi, BTK inhibitor; CR, complete response; treat as de novo DLBCL, typically R-CHOP or R-CHOP like regimens.
Proposed algorithm for the management of RT DLBCL in the era of novel agents of CLL. Bcl-2i, Bcl-2 inhibitor; BTKi, BTK inhibitor; CR, complete response; treat as de novo DLBCL, typically R-CHOP or R-CHOP like regimens.
In selected RT patients who achieve CR or PR with first-line therapy and with fit clinical status, stem cell transplantation is recommended as a consolidative approach with the goal of maintaining long-term remission.
HL-type or other types of RT
HL transformation from CLL is a recognized complication with rare occurrence.
Among 3887 CLL patients seen at the Mayo Clinic, 26 (0.7%) developed HL transformation.70 In a nested cohort of ∼2500 newly diagnosed CLL patients prospectively followed, the incidence of HL transformation was 0.05% annually. All patients in the Mayo cohort had classical HL. In contrast to the negative EBV and clonally related status in DLBCL-type RT, up to 70% of HL RT cases were EBV positive, and the majority of HL RT cases (>50%) were clonally unrelated to prior CLL.71 The median time from CLL diagnosis to HL RT development was ∼4 to 6 years. Given the rarity of this disease, no clinical trial has been performed for this cohort of patients. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD), the standard chemotherapy for de novo classical HL, is a regimen frequently used to treat HL RT. The overall response rate of ABVD in these patients ranges from 40% to 60%, and the median survival is ∼4 years.70,72,73 This outcome is worse than the overall survival of de novo HL, however appears superior to survival of DLBCL subtype of RT. Therefore, stem cell transplant was rarely performed, and only reserved for relapsed patients. In the era of novel therapy for CLL, a few scattered cases of HL RT have been reported. Of note, 3 HL RT post-Ven patients all responded to chemotherapy (ABVD, R-CHOP, or CHEP [cyclophosphamide, Adriamycin, etoposide, prednisone]), with survival >30 months currently.13 There is not enough data to recommend a different approach to manage these patients. Occasionally, post–novel agent transformation into plasmablast lymphoma7 or clonally related histiocytic sarcoma has been observed.74 Survival was short in these cases, and no clear therapy was effective.
Conclusion and perspectives
Despite advances in developing novel agents for CLL, RT continues to be a clinical area with unmet need. Because RT in the era of novel agents typically has a complex karyotype and/or TP53 disruption, preferred treatment is a clinical trial including novel agents. Critical biological questions that still need to be addressed in the era of novel agents include (1) CLL molecular evolution and risk factors in relationship to RT development, (2) heterogeneity of RT diseases after different prior therapies, (3) immune evolution in RT during CLL transformation, and (4) biomarkers that can select different RT patients for novel therapies. Given the rarity of RT in the current era of CLL novel agents, collaborative efforts from multiple academic centers are needed to answer these important questions.
Acknowledgments
The author thanks Jennifer Brown, Sameer Parikh, Nitin Jain, and Neil E. Kay, who provided critical review of this paper, and Rong He and Timothy Call, who provided important input. W.D. received support from National Cancer Institute grant K23 CA160345; Mayo Clinic Cancer Center/Richard M. Schulze Family Foundation for Awards in Cancer Research; and the Fraternal Order of Eagles Cancer Research Fund.
Correspondence
Wei Ding, Division of Hematology, Department of Internal Medicine, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: ding.wei@mayo.edu.