
Abstract
Over the past decade, there has been exponential growth in the number of genome sequencing studies performed across a spectrum of human diseases as sequencing technologies and analytic pipelines improve and costs decline. Pediatric hematologic malignancies have been no exception, with a multitude of next generation sequencing studies conducted on large cohorts of patients in recent years. These efforts have defined the mutational landscape of a number of leukemia subtypes and also identified germ-line genetic variants biologically and clinically relevant to pediatric leukemias. The findings have deepened our understanding of the biology of many childhood leukemias. Additionally, a number of recent discoveries may positively impact the care of pediatric leukemia patients through refinement of risk stratification, identification of targetable genetic lesions, and determination of risk for therapy-related toxicity. Although incredibly promising, many questions remain, including the biologic significance of identified genetic lesions and their clinical implications in the context of contemporary therapy. Importantly, the identification of germ-line mutations and variants with possible implications for members of the patient’s family raises challenging ethical questions. Here, we review emerging genomic data germane to pediatric hematologic malignancies.
Learning Objectives
Understand the genomic lesions currently used for risk stratification, targeted therapies, and individualization of chemotherapy dosing for pediatric patients with hematologic malignancies
Highlight several newly identified somatic and germ-line genetic lesions and variants with potential implications for prognostication, targeted therapeutic intervention, and determination of risk of pediatric hematologic malignancy development
Introduction
The outcomes of children with most hematologic malignancies have steadily improved over recent decades. However, certain diseases and specific subsets of patients still have suboptimal outcomes with current standard of care treatment. Additionally, standard chemotherapy can be associated with a high burden of toxicity, both immediately and lifelong, for childhood cancer survivors. These challenges have fueled the pursuit of “precision medicine” for the care of children with hematologic malignancies. As broadly defined, precision medicine includes precise assignment of patients to risk-based therapy, identification of targetable genetic lesions, and individualization of chemotherapy dosing. Recent advances have facilitated routine performance of next generation sequencing assays in clinical environments. This has facilitated the translation of genomic profiling studies of large, well-annotated cohorts of pediatric patients with hematologic malignancies being uniformly treated on clinical trials.
Here, we will review well-established and newly identified genetic lesions in pediatric hematologic malignancies. We will discuss the potential prognostic and therapeutic implications of the described somatic genetic lesions. We will also discuss germ-line genetic mutations and polymorphisms associated with childhood leukemia risk and chemotherapy-induced toxicities.
B-lymphoblastic leukemia
Recurrent somatic genetic lesions are an integral component of risk stratification algorithms for pediatric B-lymphoblastic leukemia (B-ALL) for most large pediatric cancer consortia (Table 1). The majority of these lesions are structural chromosomal alterations that are associated with the development of disease and have prognostic implications.
Recurrent structural chromosomal aberrations in B-ALL
Hyperdiploidy (modal chromosome numbers 51-65 or DNA index of >1.16) is common in B-ALL, occurring in 20% to 25% of pediatric patients and decreasing in frequency with increasing age. Patients with hyperdiploidy generally do well, with studies from the Children’s Oncology Group (COG) finding that specific trisomies (trisomy of chromosomes 4 and 10) in particular are linked to a favorable outcome1 (Table 1).
Conversely, hypodiploidy with modal chromosome number <44 or DNA index of <0.81 has been associated with a dismal outcome, resulting in hematopoietic stem cell transplant (HSCT) in first complete remission (CR).2 However, recent data from a small series of patients treated at a single institution suggest that, if a patient with hypodiploidy has a bone marrow that is negative for minimal residual disease (MRD) by the end of induction therapy, chemotherapy alone may be curative.3 Recently analyzed data from the COG indicate that, although patients with an end of induction MRD < 0.01% fare better than those with MRD-positive disease, their overall outcomes with chemotherapy alone were still suboptimal, with a 5-year disease-free survival of 60.3% ± 9.2%. However, this study found that HSCT in first CR did not confer a survival benefit, suggesting a need for the development of novel therapeutic strategies for this poor prognosis subset.4
In addition to aneuploidy, a number of chromosomal translocations and other structural chromosomal aberrations with prognostic impact are common in B-ALL. The most common translocation in pediatric B-ALL is t(12;21), which results in the ETV6-RUNX1 fusion gene. ETV6-RUNX1 fusions are present in 20% to 25% of pediatric B-ALL cases, and they are associated with an excellent outcome.5 Conversely, rearrangements involving the KMT2A gene (formerly MLL) on chromosome 11q23 are associated with higher levels of residual disease at the end of induction, which when present, are associated with inferior outcomes. Approximately 80% of infants with B-ALL have KMT2A-r compared with 3% to 5% in older children with B-ALL. Whereas infants with KMT2A rearrangements have a dismal prognosis,6 older patients have a more favorable outcome with appropriate intensification of therapy.7 Additionally, intrachromosomal amplification of chromosome 21 is a recurrent lesion in 1% to 3% of pediatric B-ALL cases that is associated with increased risk of relapse. However, treating affected patients with intensification of chemotherapy can mitigate this risk.7-9 Also, B-ALL harboring the translocation t(17;19), which results in the TCF3-HLF fusion gene, is characterized by hypercalcemia, coagulopathy, and a dismal outcome. Although rare (<1% of B-ALL patients), the recognition of t(17;19) is critical, because such patients warrant early consideration of HSCT and are candidates for novel agents—recent preclinical data suggest that these patients may be sensitive to BCL-2 inhibition.10
Rearrangements involving monocyte enhancer factor D2 (MEF2D) occur in 3% to 4% of pediatric B-ALL and tend to occur in older children.11 MEF2D rearrangements result in fusion with multiple partners, all leading to similar gene expression profiles, including overexpression of the MEF2D target HDAC9, and implicating HDAC inhibition as a potential therapeutic strategy for MEF2D-rearranged patients. Retrospective studies of small cohorts of patients have found an association between MEF2D fusions and inferior outcome, although prospective studies of larger cohorts are needed.11,12
Additionally, rearrangements of the zinc finger protein 384 gene (ZNF384) have been identified in 4% to 5% of pediatric B-ALL patients.13,14 These rearrangements lead to the fusion of ZNF384 with multiple partners, including TCF3, EWS1, CREBBP, and EP300.13,14 ZNF384-rearranged B-ALL is characterized by weak CD10 expression and aberrant expression of myeloid markers and a gene expression signature enriched for stem cell–related genes.13 Based on small cohorts thus far examined, it seems that the outcomes of patients with ZNF384 fusions are not statistically different than those without, although fusion partner-specific effects may exist. Hirabayashi et al13 reported that patients with TCF3-ZNF384 fusions were characterized by younger age at diagnosis, relative resistance to corticosteroids, and higher risk of relapse. Additional studies are needed to definitively determine the impact on outcome of these fusions.
Philadelphia chromosome and Philadelphia chromosome–like B-ALL
Perhaps the best characterized structural chromosomal alteration of B-ALL is the t(9;22) Philadelphia chromosome (Ph+), leading to the BCR-ABL1 fusion gene.15 Although historically associated with a very poor outcome, incorporation of continuously administered tyrosine kinase inhibitors (TKIs) targeting BCR-ABL1, such as imatinib and dasatinib, into standard chemotherapy without HSCT has dramatically improved outcomes of Ph+ B-ALL patients in recent years and is now the well-accepted standard of care treatment of this disease.16-18
Global gene expression analyses of B-ALL cohorts identified a group of patients whose disease had a gene expression profile largely overlapping with Ph+ B-ALL, despite lacking the BCR-ABL1 fusion gene. Such cases are classified as Ph-like B-ALL (or BCR-ABL1–like B-ALL), and as a group, they have an extremely poor prognosis. Ph-like disease is common in high-risk B-ALL, including in over 20% of National Cancer Institute (NCI) high-risk patients19,20 (Figure 1). The incidence of Ph-like disease increases with age from ∼10% in NCI standard-risk patients to over 20% of adolescents, with an apparent peak in young adults with B-ALL, over 30% of whom will have Ph-like disease.20-22
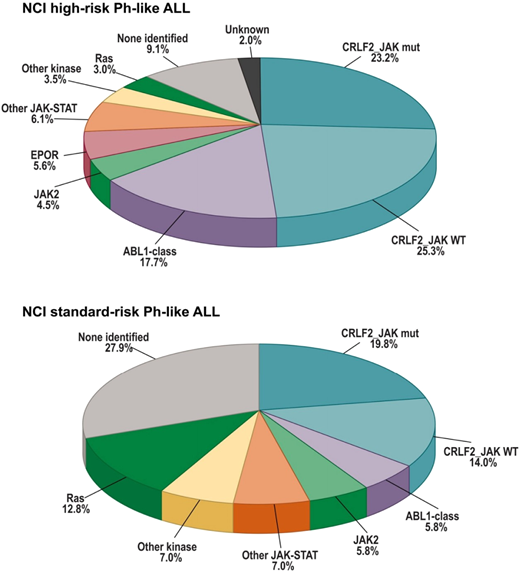
Frequencies of kinase subgroups in NCI high-risk (HR) and standard-risk (SR) patients who are Ph like. Reprinted from Reshmi et al20 with permission.
Frequencies of kinase subgroups in NCI high-risk (HR) and standard-risk (SR) patients who are Ph like. Reprinted from Reshmi et al20 with permission.
Extensive genomic studies have now identified a number of lesions driving Ph-like B-ALL, comprising a host of genomic alterations and leading to activation of kinase signaling pathways.20-27 Importantly, many of the identified kinase fusions and mutations are targetable by clinically available small molecule inhibitors of tyrosine kinases, offering hope that targeted therapy may improve outcome in affected patients akin to the improved outcomes in Ph+ acute lymphoblastic leukemia (ALL).
Approximately 1/2 of all Ph-like ALL cases harbor lesions leading to oncogenic overexpression of the CLRF2 gene, which encodes the thymic stromal lymphopoietin receptor that, together with the interleukin-7 receptor (IL7R), activates the JAK/STAT pathway on ligand binding (Figure 1). Therefore, overexpression of CRLF2 leads to aberrant hyperactivation of the JAK/STAT pathway. Curiously, ∼1/2 of CRLF2-overexpressing B-ALLs also have a concomitant activating mutation of JAK1 or JAK2. Additionally, rearrangements of the erythropoietin receptor (EPOR) that generate a truncated protein lacking the negative regulatory domain drive a small subset of Ph-like B-ALL via constitutive activation of the JAK/STAT signaling pathway.28 These, along with a handful of other JAK/STAT-activating lesions (including JAK2 fusions, IL7R insertion/deletions and mutations, and SH2B3 deletions), point toward the JAK pathway as an attractive potential target for the treatment of this prominent subset of Ph-like B-ALL. A number of clinical trials including pediatric patients are currently ongoing to assess the safety, tolerability, and efficacy of the combination of the JAK1/2 inhibitor, ruxolitinib, with standard chemotherapy for JAK-activated Ph-like B-ALL (NCT02723994, NCT03117751, and NCT02420717).
Additionally, a number of kinase fusions leading to activation of ABL class kinases, such as ABL1, ABL2, CSF1R, PDGFRA/B, and LYN, occur in 3% to 5% of pediatric B-ALL cases.20-22 Such fusions are predicted to be targetable by ABL kinase inhibitors, such as imatinib and dasatinib. Given the known safety and tolerability of these TKIs administered continuously on a standard intensified chemotherapy backbone, a number of clinical trial groups are now studying the efficacy of continuous dasatinib plus standard chemotherapy for Ph-like patients with ABL class lesions (NCT02883049, NCT03117751, and NCT02420717). Lesions of other activating signaling kinases, such as NTRK3, FLT3, and FGFR1, are rare but represent additional potentially targetable lesions in Ph-like ALL. Promising preclinical data and anecdotal reports of patient responses to TKI therapy provide hope that a precision medicine approach to Ph-like B-ALL will ultimately improve the outcome of this clinically challenging subset of patients.
Other secondary genetic lesions in B-ALL
A boon of recent genomic studies has greatly expanded our view of the genomic landscape of B-ALL (Table 1). Such studies have identified a number of recurrent genomic lesions with potential prognostic and therapeutic implications. Analyses of DNA copy neutral alteration by single-nucleotide polymorphism array studies have identified recurrent deletion of several transcription factors critical to normal B-cell development, including PAX5, EBF1, and IKZF1.29 Although deletion of each of these genes likely contributes to the development of B-ALL, only IKZF1 deletion seems to impact prognosis, with IKZF1 deletions and mutations being associated with a poor prognosis.30-32 The prognostic strength of IKZF1 status can be further refined by integration of additional genomic lesions. In a large study of patients treated on the Associazione Italiana Ematologia ed Oncologia Pediatrica-Berlin-Frankfurt-Muenster ALL 2000 trial, patients with IKZF1 deletion and concomitant deletion of CDKN2A, CKN2B, PAR1, or PAX5 without deletion of ERG (termed IKZF1plus) had a significantly worse 5-year event-free survival (EFS) than those with IKZF1 deletions without the IKZF1plus status, and those without IKZF1 deletions (53% ± 6% vs 79% ± 5% vs 87% ± 1%, respectively).33 IKZF1 lesions are highly enriched in high-risk subsets of B-ALL, including over 60% of Ph+ ALL and high frequency in Ph-like B-ALL, which suggests a specific interaction with these activating kinase lesions.19,30,34,35 Work to investigate the features of IKZF1 lesions that lead to a poor prognosis has found that loss of IKAROS (the protein encoded by IKZF1) function leads to a stem cell–like gene expression signature and increased adhesion to the bone marrow niche. In addition to potential use as a prognostic marker, preclinical work in Ph+ ALL has identified potential targeted therapeutic interventions for B-ALL with IKZF1 lesions. The efficacy of retinoids as well as pharmacologic inhibition of the FAK kinase in combination with TKIs has been shown in preclinical studies of IKZF1-deleted/mutated Ph+ B-ALL.36,37
More recently identified recurrent genomic lesions in B-ALL with distinct transcriptional signatures and potential prognostic impact include combined deregulation of DUX4, a homeobox transcription factor, and ERG, a member of the erythroblast transforming–specific transcription factor family. These occur in 7% of pediatric B-ALL, and they are associated with favorable outcome.38 Interestingly, DUX4-ERG–dysregulated B-ALL frequently co-occurs with deletions of IKZF1, but unlike in other B-ALL subtypes, concomitant IKZF1 deletion does not seem to negatively impact outcome.38
T-cell ALL
In contrast to B-ALL, recurrent genomic lesions are not widely incorporated into the risk stratification schemas for children with T-cell acute lymphoblastic leukemia (T-ALL) (Table 2). Clinical data suggest limited impact of any given genomic lesion on outcome with modern therapy. Therefore, current risk stratification for therapy generally relies on clinical features, such as extramedullary involvement and early response to induction therapy. Indeed, even white blood cell count and age at diagnosis, known to be prognostically significant for B-ALL, are not considered for patients with T-ALL. However, integration of genetic information and MRD for risk stratification is being explored, and potentially targetable genetic lesions are common; thus, possible future applications for genomics of T-ALL are on the horizon.
Recurrent somatic genetic lesions
Structural chromosomal aberrations are not prognostic in T-ALL, including rearrangements involving TAL1, KMT2A, MLLT10, TLX1, and TLX3. Deletions of CDKN2A/B and PTEN are present in ∼50% and ∼20%, respectively. The most commonly mutated gene in T-ALL is the transmembrane receptor involved in normal T-cell development, NOTCH1, present in >70% of patients. Mutations of the ubiquitin ligase gene, FBXW7, activated signaling pathways, and epigenetic regulators are also common.39
Efforts to incorporate genetic lesions to refine risk stratification are being explored. One example is a recent study by the pediatric French Acute Lymphoblastic Leukemia Study Group.40
The researchers defined patients as genetic low risk (gLoR) if they had a mutation of either NOTCH1 or FBXW7 and lacked mutations of the Ras pathway and PTEN. All other patients were classified as genetic high risk (gHiR). Combining genetic risk and end of induction MRD at a threshold of 10−4 allowed for a refinement of risk stratification, distinguishing a group with an excellent outcome (gLoR: MRD < 10−4 and low cumulative risk of relapse [CIR] =4%) from a group with a very poor expected outcome (gHiR: MRD > 10−4, CIR = 43%). Other patient groups (gLoR: MRD > 10−4 and gHiR: MRD < 10−4) had intermediate outcomes.40 Thus, if these results are validated in additional cohorts, adopting an approach integrating clinical features, genetic lesions, and early response to therapy may allow for improved risk stratification of T-ALL.40
The frequency of activating NOTCH1 mutations and loss of function mutations of the negative NOTCH1 regulator, FBXW7, in T-ALL makes the NOTCH1 signaling pathway an attractive potential therapeutic target. Targeting NOTCH1 signaling using γ-secretase inhibitors that prevent release of the transcriptional activating intracellular portion of NOTCH1 from the membrane has shown promising preclinical activity but thus far, limited clinical efficacy and high rates of dose-limiting gastrointestinal toxicity.41 NOTCH1 inhibitory antibodies are also being explored as an NOTCH1 targeting strategy. Additionally, NOTCH1 activation directly upregulates MYC; thus, targeting MYC may be an effective strategy for T-ALL with NOTCH1 activation.41
Early thymic precursor ALL
Early thymic precursor or early T-cell precursor (ETP)-ALL is a distinct subtype of T-ALL characterized by a stem progenitor cell gene expression signature and an immature immunophenotype. Initial reports found that pediatric patients with ETP-ALL were a high-risk subset associated with poor response to therapy and high risk of relapse.42 More recent pediatric data suggest that, despite having higher rates of MRD positivity at early time points, with MRD-directed intensification of therapy, outcomes are not significantly different than typical T-ALL.43-45 Therefore, ETP status is currently not universally incorporated into risk stratification for pediatric T-ALL.
Although not independently prognostic, the requisite for intensive therapy for cure in ETP-ALL indicates that this subset of patients could benefit from more targeted approaches to therapy. The molecular landscape of ETP-ALL reveals a number of potentially targetable opportunities, including mutations of activated signaling pathways.38
Acute myeloid leukemia
Recurrent structural chromosomal and molecular lesions are the most powerful prognostic predictors for patients with acute myeloid leukemia (AML) (Table 3). However, current risk stratification of pediatric AML takes into consideration only a limited number of somatic genetic lesions, many of which, although common in adult AML, affect only a small fraction of pediatric patients.46,47 Therefore, the majority of pediatric patients lack any prognostic genetic lesion. Recent genome-sequencing efforts of large cohorts of pediatric AML cases, including the Therapeutically Applicable Research to Generate Effective Treatments AML initiative and the St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project, have identified genetic lesions enriched in pediatric AML, including novel fusion genes, mutations, and deletions that may have important clinical implications specific to pediatric AML.46,47
Structural chromosomal aberrations in AML
Structural chromosomal aberrations creating fusion genes affecting the hematopoiesis-regulating transcription factor core binding factor complex are present in ∼25% of pediatric AML. The α subunit of this transcription factor complex, RUNX1, is fused to the RUNX1T1 gene in t(8;21) AML, whereas the β cofactor subunit of the complex, CBFb, is fused to MHY11 in inv(16) or t(16;16) AML. Collectively, these lesions are associated with a relatively favorable outcome, and therefore, they are not routinely offered HSCT in first CR. Conversely, the recurrent structural chromosomal aberrations of monosomy 5, 5q- and monosomy 7 are associated with particularly poor outcomes, with HSCT in first CR a generally accepted standard of care, although such lesions are quite uncommon in pediatric patients.48 Complex karyotypes (≥3 structural chromosomal lesions) are relatively common in pediatric AML; however, in contrast to adult disease, they are not correlated with outcome, and therefore, they are not routinely incorporated into risk classification for children.48 Another set of well-characterized structural chromosomal lesions in AML is lesions of 11q23 leading to fusion of the KMT2A gene with a host of different partners. Although the prognostic impact of KMT2A-r as a group has not been consistently shown , clinical data suggest that certain KMT2A rearrangements, such as t(10;11)(p11.2;q23) and t(6;11)(q27;q23), may portend a particularly poor prognosis, whereas the t(1;11)(q21;q23) translocation has been associated with an excellent outcome.49
A number of recent studies have identified recurrent cryptic translocations involving the nucleoporin 98-kDa (NUP98) gene on chromosome 11p15 in AML in ∼4% of pediatric AML patients, with decreasing frequency with increasing age.50 Although the most common fusion partner is the histone methyltransferase gene, NSD1, over 30 different fusion partners have been identified to date. NUP98-rearranged AML occurs almost exclusively in cytogenetically normal AML with frequent concomitant FLT3-internal tandem duplication (-ITD) and WT1 mutations and is characterized by aberrant HOX gene expression.46,50-52 Several retrospective studies have found that patients with NUP98 rearrangement have a particularly poor outcome, suggesting the possible utility of incorporating NUP98 status into risk classification.50-52 However, as discussed below, the constellation of co-occurring mutations likely drives the prognostic impact rather than NUP98 status considered in isolation.46,50
Acute promyelocytic leukemia
Specific fusions involving the retinoic acid receptor α (RARA) due to balanced translocations, most commonly, t(15;17)(q22;q12) producing PML-RARA and very rarely, fusions involving retinoic acid receptor β and retinoic acid receptor γ, cause acute promyelocytic leukemia (APL), a clinically and biologically distinct form of AML.53,54 Patients with APL generally have an excellent outcome, but they have relatively high rates of early death due to severe coagulopathy and differentiation syndrome. The RARA fusion genes result in the production of an abnormal retinoid acid receptor that causes the repression of RARA target genes, leading to blocked differentiation at the promyelocytic stage. This differentiation block can be overcome by treatment with all-trans-retinoic acid (ATRA), and treatment regimens combining ATRA and standard AML-directed chemotherapy are associated with excellent outcomes in APL, with CR rates up to 95% and survival >80%. Additionally, arsenic trioxide is a particularly active agent in APL, thought to work by binding to the PML portion of the PML-RARA fusion and causing its degradation. Adult studies have shown that, for low- and standard-risk APL patients, the combination of ATRA and arsenic without chemotherapy is as efficacious as standard chemotherapy but with much less toxicity.55 Studies of this combination for the upfront treatment of pediatric APL patients are ongoing (NCT02339740 and NCT01409161).
Somatic molecular lesions in AML
One of the most commonly mutated genes in AML is the receptor tyrosine kinase gene, FLT3. ITD mutations of the juxtamembrane domain (JMD) and point mutations of Asp835 in the tyrosine kinase domain (TKD) are the most common mutations of FLT3 in AML. FLT3-TKD mutations occur in around 10% of adult and pediatric AML patients, whereas FLT3-ITD mutations are far more common in adults, present in ∼35% compared with only 10% to 15% of pediatric patients.56 Although FLT3-TKD mutations do not seem to significantly impact prognosis, dominant FLT3-ITD lesions, as evidenced by an allelic ratio >0.4, are associated with a poor outcome.56
Patients with high allelic ratio FLT3-ITD lesions are generally categorized as high-risk patients, warranting HSCT in first CR. In addition to its prognostic implications, as a receptor tyrosine kinase, FLT3 is potentially targetable by small molecule inhibitors.57 A number have been used in clinical trials, including sorafenib in a current AML trial recently run by the COG (NCT01371981). Adult studies looking at other TKIs targeting FLT3 have had promising results, most prominently the improved overall survival and EFS observed in adults with FLT3-mutated AML treated with the multikinase inhibitor midostaurin, which is now Food and Drug Administration approved for this use.58,59 Resistance to TKIs can arise through a variety of cell intrinsic and extrinsic mechanisms, including the emergence of resistance-conferring mutations of the TKD of FLT3.60,61 Many of the commonly occurring TKD mutations remain sensitive to some of the newer TKIs, such as crenolanib and gilteritinib, and therefore, they could be efficacious for patients in whom resistance to first generation TKIs has developed.62,63 Although FLT3-ITD mutations are less frequent in pediatric AML patients compared with adult AML patients, pediatric-specific FLT3 mutations have been recently identified. Novel point mutations and small insertions/deletions of not only the TKD but also, the JMD and trans-membrane domain have been identified in children.46,64 These mutations are associated with poor response to standard therapy but display exquisite sensitivity to FLT3-targeting TKIs in in vitro studies.46,64 Thus, identification of such mutations at diagnosis could inform risk stratification and point toward targeted therapeutic intervention.46,64
In contrast to FLT3-ITD mutations, recurrent mutations of NPM1 and CEBPA genes are associated with a favorable outcome in pediatric AML.65,66 Mutations of NPM1 constitute one of the most frequent mutations in adult AML but are relatively rare in pediatric disease, occurring in <10% of patients.46,65 CEBPA mutations are also uncommon in pediatric AML, found in ∼5% of patients, and they are more common in older children compared with infant and young children.46,66 Both NPM1-mutant and CEBPA-mutant AML are now provisional entities in the World Health Organization classification of myeloid malignancies. Although rare, routine screening for NPM1 and CEBPA mutations is recommended, because such patients are candidates for treatment with chemotherapy alone. Additionally, novel therapeutic strategies for the treatment of NPM1-mutant AML are being explored. Mutant NPM1 seems to disrupt normal chromatin structure, leading to aberrant HOX gene expression, and targeting of histone modifications, including DOT1L inhibitors and inhibitors of the menin-KMT2A interaction, has shown promise in preclinical investigations.67 Interestingly, a small clinical trial in adults with NPM1-mutant AML showed dactinomycin as a promising agent, likely working because NPM1-mutant cells are more vulnerable to the dactinomycin-induced nucleolar stress response.68
Other recurrent mutations in pediatric AML include mutations of the zinc finger transcription factor gene, WT1. Mutations of this gene are found in around 10% of children. Although patients with WT1 mutations have a worse EFS and overall survival, when combined with FLT3 status and cytogenetics, WT1 status has no clear prognostic impact.69 Additionally, a number of molecular lesions common in adult AML, including mutations of epigenetic modifiers DNMT3A, TET2, and IDH1/2, are rare in pediatric disease.46,70,71 Other significant differences in the frequency and spectrum of mutations of genes, such as WT1, MYC, GATA2, CBL, NRAS, and KRAS, exist between adult and pediatric AML, including a number of mutations exclusive to pediatric disease.46 Focal deletions of ELF1, ZEB2, and MBNL1 genes have also been identified predominantly in pediatric cases.46 Defining the prognostic impact of these pediatric-specific lesions could ultimately help refine pediatric AML risk classification and point toward potential pharmacologically targetable lesions.46
An integrated genetic approach to risk stratification of pediatric AML
Consideration of combinations of mutations seems to more precisely define prognostic subgroups in AML rather than consideration of individual lesions in isolation.46 Evaluation of multiple cohorts of pediatric AML patients found that those with both an FLT3-ITD and concomitant NPM1 mutation had a particularly favorable outcome, despite numerous prior studies showing a negative impact of FLT3-ITD mutations alone in AML. Conversely, patients with an FLT3-ITD and either a WT1 or NUP98-NSD1 fusion had considerably worse outcomes than those patients with FLT3-ITD mutation alone.46 Thus, additional analysis of large, well-annotated pediatric cohorts could establish a pediatric-specific genomic classification system akin to those used for adult disease.47,72 Of course, the potential tradeoff for such precision is complexity. Pediatric AML in general is a rare disease, and if divided into small subgroups of genomically identified patients, the power to detect significant differences in outcome could be lost. Ultimately, perhaps the most practical means to adequately risk stratify pediatric AML patients will be integration of cytogenetic and molecular risk for group and MRD determination.73,74 This will allow for the further refinement of classification for patients with well-defined genetic classifiers and an opportunity to risk stratify the large group of patients lacking such classifiers.46
Acute megakaryoblastic leukemia
Focused sequencing of specific AML subsets common in pediatric AML has identified distinct genomic subgroups with prognostic impact. Perhaps the best example of this is in acute megakaryoblastic leukemia (AMKL), a rare disease in adults (<1% adult AML75 ) but relatively common among pediatric patients (60%-90% of pediatric Down syndrome [DS] AML76,77 and 5%-7% of pediatric non-DS AML78,79 ). DS-AMKL has a favorable outcome, as most such patients are curable with relatively low-intensity chemotherapy.80-83 Conversely, AMKL in non-DS patients is often associated with a poor prognosis. RNA sequencing and exome sequencing of a large cohort of non-DS AMKL patients identified genetic subgroups with prognostic impact.84 Patients with CBFA2T3-GLIS2 fusions had a dismal prognosis, with an overall survival of 14%-16%, whereas remarkably, no patients with GATA1 mutations relapsed.84-86 Patients with KMT2A rearrangements and NUP98-KDM5A fusions also did poorly, whereas those with HOX gene rearrangements and RMB15-MKL1 fusions had an intermediate prognosis.84 It was recently discovered that CBFA2T3-GLIS2 fusions AMKL is characterized by the overexpression of a number of Hedgehog-related genes and that targeting of this pathway using inhibitors of the downstream effectors of the Hedgehog pathway, GLI, could be an effective therapeutic strategy for this extremely poor prognosis subset.87
Genetic lesions recurrent across a spectrum of pediatric hematologic malignancies
Although many disease-specific genetic lesions characterize pediatric hematologic malignancies, a handful of lesions occur across multiple disease types, and thus, effectively targeting such lesions could have a broad impact on childhood leukemia.
The Ras/MAPK pathway is perhaps the most commonly mutated pathway in pediatric hematologic malignancies and human cancer in general. Ras-activating mutations are the genetic hallmark of juvenile myelomonocytic leukemia,88 but they also commonly occur in subsets of lymphoid and other myeloid malignancies. Ras pathway mutations occur in a spectrum of B-ALL subtypes and are particularly common in near-haploid B-ALL in patients with DS and high-hyperdiploid ALL.89-92 Activating RAS mutations are enriched at B-ALL relapse, suggesting that they may confer resistance to standard chemotherapy.90,93 Furthermore, around 15% of pediatric T-ALL patients harbor Ras pathway mutations,39 and mutations of the Ras pathway are common in pediatric AML, particularly in children <3 years of age.46 Although potentially promising, targeting Ras has proven difficult. Initially believed to be “undruggable,” direct Ras-targeting small molecules have recently shown promising preclinical activity.94,95 Most recent efforts have instead focused on targeting the downstream MAPK and PI3K signaling pathways. MEK and PI3K inhibitors are in various stages of clinical development for relapsed/refractory pediatric leukemias.
Rearrangements of KMT2A also occur across a spectrum of pediatric hematologic malignancies, including over 75% of infant ALL. KMT2A-r infant ALL is associated with a dismal outcome.6 KMT2A rearrangements have also been associated with a poor prognosis in noninfant B-ALL,7,96 T-ALL,91 and AML49,84 and are common in the poor prognosis mixed phenotype acute leukemia and therapy-related AML.97 Thus, effective targeted therapy for KMT2A-rearranged leukemia is a critical need. Global gene expression analysis of KMT2A-r ALL identified overexpression of FLT3, and preclinical work suggested potential efficacy of FLT3 inhibition in KMT2A-r ALL98 ; however, a trial combining the FLT3 inhibitor lestaurtinib failed to show a survival benefit in infant KMT2A-rearranged ALL.99 KMT2A-r leukemia is characterized by potentially targetable epigenomic aberrations as well. The histone methyltransferase, DOT1L, was shown to be critical to KMT2A-r leukemias, and preclinical data of small molecule inhibitors of DOT1L generated tremendous enthusiasm.100,101 However, a phase 1 study of the DOT1L inhibitor, pinometostat (EPZ5676), showed good tolerability but failed to show an efficacy signal in pediatric KMT2A-r leukemia patients.102 Additional strategies targeting the epigenetic aberrations that underlie KMT2A-r leukemia are being investigated, including agents targeting the menin-KMT2A interaction,103 the arginine methyltransferase PMRT5,104 and the histone demethylase LSD1.105 KMT2A-r leukemias are also characterized by aberrant DNA hypermethylation; thus, incorporation of the hypomethylating agent azacitidine into standard chemotherapy is being explored in a clinical trial of infant KMT2A-r leukemia (NCT02828358).
Genomics of relapse
Genomic studies of matched diagnostic, remission, and relapse samples have allowed for exploration of the clonal dynamics of pediatric leukemias. At diagnosis, there is significant genetic heterogeneity with clonal evolution throughout disease progression. These important studies have identified specific genetic lesions that are specific to or enriched at relapse. Such lesions likely confer resistance to chemotherapy, allowing for the survival of a subclone that then expands and acquires new mutations to drive disease relapse.
Mutations of the gene, NT5C2, are among the most commonly occurring relapse-specific mutations in ALL. NT5C2 is a 5′-nucleotidase important in maintaining intracellular nucleotide levels. The mutations occurring in ALL are gain of function mutations, producing an activated NT5C2 protein that leads to 6-mercaptopurine (6-MP) and 6-thioguanine resistance through the inactivation of the purine nucleoside analogs.106 NT5C2 mutations are frequent in both relapsed B-ALL and T-ALL, with all patients harboring such mutations relapsing within 36 months of diagnosis.107,108 Screening for the emergence of NT5C2 mutations could identify patients who would benefit from an altered therapeutic approach, such as the introduction of additional noncross-reactive chemotherapeutic agents during maintenance therapy.109 Additionally, targeted therapeutic intervention may be possible, with inhibitors of NT5C2 being explored and data to support the use of inosine-5′-monophosphate dehydrogenase inhibition to mitigate the chemoresistance induced by NT5C2 mutations.106,110
Additional relapse-enriched mutations in ALL have been identified, and the mechanisms by which they emerge under the selective pressure of chemotherapy are being elucidated. For example, mutations of the epigenetic regulator CREBBP are common at relapse in pediatric ALL.90,93,111,112 CREBBP encodes histone acetyltransferase CREB binding protein, a transcriptional coactivator. The genetic lesions that occur in ALL are loss of function mutations or deletions, which lead to aberrant transcriptional regulation of CREBBP targets, including glucocorticoid response elements. Thus, these mutations likely contribute to relapse via resistance to glucocorticoid therapy.90,112 Mutations of a number of additional epigenetic regulators are highly enriched at relapse in ALL, including SETD2, WHSC1, KMT2D, EP300, KDM6A, and MSH6, strongly implicating epigenetic dysregulation as a key mediator of resistance to chemotherapy.93,111,113 The frequency of such mutations also points toward the possibility of incorporating epigenetically targeted agents as a means to overcome chemoresistance in ALL. Additionally, deletions and mutation of IKZF1 are enriched at relapse along with activating Ras pathway mutations.90,93,111
Likewise, in AML, selective pressure of chemotherapy leads to clonal evolution.114-117 Elegant work in adult AML revealed that mutations in genes involved in regulation of global chromatin structure, such as DNA methylation regulators, histone modifiers, and chromatin looping, arise in preleukemic stem/progenitor cells with malignant transformation after the acquisition of additional cooperative mutations. These preleukemic stem cells often survive chemotherapy and serve as reservoirs for relapse.114 The evolution of pediatric AML is likely different, because the founding epigenetic modifier mutations common in adult AML are rare in pediatric disease. However, sequencing studies of relapsed pediatric disease have identified relapse-enriched mutations of epigenetic regulators, such as CREBBP, SETD2, and ASXL3, indicating a role for epigenetic aberration in the persistence of a relapse-initiating clone in pediatric AML as well.115,116 Additionally, signaling mutations, including activating PTEN, NRAS, and TKD mutations of FLT3, seem to be enriched at relapse in pediatric AML and may represent targetable lesions, although larger studies are warranted.116,117
Germ-line genetic variation in pediatric hematologic malignancies
Pharmacogenomics
Tolerance of standard chemotherapeutic agents can vary substantially. Recent work has uncovered a number of genetic variants in pediatric leukemia patients associated with poor tolerance of specific chemotherapies and increased risk for certain chemotherapy-induced toxicities. Such studies lay the groundwork for individualized administration of chemotherapeutic agents.
The antimetabolite 6-MP is a critical component of therapy for ALL, but vast interindividual tolerance exists. Polymorphisms of the thiopurine metabolism gene, thiopurine S-methyltransferase (TMPT), are a well-established risk factor for 6-MP toxicity,118 and more recently, identified coding variants of the nudix hydrolase 15 (NUDT15) gene are also highly associated with increased 6-MP–induced hematologic toxicity.119 It is now generally accepted routine practice to screen all ALL patients for the TMPT genotype, and newer guidelines have been established to screen individual in the highest-risk ethnic groups for NUDT15 toxicity–associated variants and dose adjust 6-MP accordingly.119 This type of individualized dosing should reduce toxicity and minimize the associated delays in therapy.
Germ-line polymorphisms associated with asparaginase toxicities, vincristine neuropathy, and corticosteroid-induced osteonecrosis have also been identified through recent genome-wide association studies (GWAS) of pediatric leukemia patients.120-123 The clinical application of these polymorphisms is less well defined than for the 6-MP metabolism–altering variants. However, one can envision that the prospective identification of these risk alleles could be used to identify patients in need of closer monitoring for specific toxicities and tailor enhanced supportive care measures in individuals with toxicity-associated polymorphisms.
Genetic susceptibility
Although cancer predisposition syndromes are thought to be rare underlying causes of childhood hematologic malignancies, recent studies have identified some important exceptions. Sequencing of paired tumor and germ-line material from pediatric patients with hypodiploid B-ALL identified mutation of TP53 in over 90% of B-ALL patients with low hypodiploid (32-39 chromosomes), 50% of which were present in the germ line.89 This seminal finding has important clinical and potential ethical implications. As a high-risk subset of B-ALL, such patients are frequently offered HSCT in first CR. The presence of a familial germ-line TP53 mutation in a matched sibling would make them an unsuitable donor, necessitating screening of siblings. Additionally, given the high risk of cancer development in patients with germ-line TP53 mutations, surveillance guidelines exist. Therefore, screening of other family members is advisable. Additionally, rare germ-line mutations in genes, such as ETV6 and PAX5, have been identified in familial as well as sporadic B-ALL.124-127 Similarly, germ-line GATA2 mutations have recently been identified in a high percentage of adolescent patients with myelodysplastic syndrome (MDS) with monosomy 7.128 Screening of potential sibling donors and other family members would also need to be discussed with the families of affected patients. Additional MDS/AML predisposition germ-line mutations are also well described, including ANKRD26, CEBPA, DDX41, ETV6, RUNX1, SRP72, SAMD9, and SAMD9L genes.129 Thus, kindred with histories of multiple family members with childhood- and young adult–onset hematologic malignancies should be referred for genetic screening.130,131 This raises a number of potential issues for affected families, including implications for the genetic testing and medical care of siblings and parents, the psychological impact of cancer surveillance, and reproductive planning regarding risk for future offspring. Additionally, families may feel pressure to contact estranged relatives, and genetic test results may reveal the presence of the mutation in a family member who wishes not to know their status. Furthermore, in the event of a patient’s death, reporting of results is complex.132 Thus, in addition to the medical challenges that they are facing, genetic testing potentially raises social, psychological, and ethical issues for affected families.124-127,131
In addition to these bona fide cancer predisposition syndromes, a number of recent GWAS of childhood leukemia have identified a number of germ-line genetic variants highly associated with risk of pediatric ALL, including common variants in genes such as ARID5B, CEBPE, and IKZF1. Additionally, GATA3 germ-line variants are specifically associated with adolescent and young adult (AYA) Ph-like and non–Ph-like B-ALL.133,134 These studies have dramatically expanded our view of the biology of ALL development but also raise important ethical questions. Are there patients who should be screened for these germ-line variants, particularly subsets strongly linked to a particular risk allele, like AYA patients with Ph-like disease? Patients and their parents often ask if other family members are at risk or if the risk of leukemia development is something that they may pass on to their children. We now know that, in a portion of cases, potentially heritable risk does exist; therefore, should we seek to identify families carrying these risk alleles? Given the lack of certainty, routine screening of patients and families is not currently recommended, but with more genomic data emerging, these questions must be considered. Referral to a cancer genetics clinic should be considered for families with multiple cases of childhood leukemia.130,131
In conclusion, genetic profiling of pediatric hematologic malignancies has led to improved risk stratification and identified a number of potentially targetable genetic vulnerabilities. Such efforts have translated into improved outcomes in specific subsets of patients (eg, Ph+ B-ALL patients treated with TKIs plus chemotherapy), with great hope for similar improvements in others (eg, Ph-like B-ALL). Interrogating the germ line of patients and unaffected controls has identified risk alleles for the development of pediatric leukemias as well as variants associated with differing susceptibility to a number of chemotherapeutic agents. Although these successes rightly generate enthusiasm, much work remains. The prognostic impact of genetic lesions identified in retrospective cohorts must be validated in patients treated with modern era therapy and integrated into the existing risk stratification schemas. Identified potentially targetable lesions must be validated as true molecular dependencies, and the safety and efficacy of incorporating targeted therapies into standard chemotherapy backbones will need to be determined. Additionally, although therapeutic effect is ideally determined through the conduct of appropriately powered clinical trials, this will not be possible for potentially targetable genomic lesions that occur in rare subsets of patients. Thus, we will likely need to reconsider how we test for and define efficacy of specific targeted agents. The growing number of genetic variants associated with increased risk of pediatric hematologic malignancies raises clinical and ethical questions, which will need to be addressed by pediatric oncologists, geneticists, and genetic counselors. Although formidable, taken together, these challenges are worth tackling given the large potential payout of improved outcomes for pediatric leukemia patients.
Correspondence
Mignon L. Loh, Department of Pediatrics, Benioff Children’s Hospital and Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, 1450 3rd St, San Francisco, CA 94158; e-mail: mignon.loh@ucsf.edu.
