Abstract
The α-thalassemia trait, associated with deletions removing both α-globin genes from 1 chromosome (genotype ζ αα/ζ--), is common throughout Southeast Asia. Consequently, many pregnancies in couples of Southeast Asian origin carry a 1 in 4 risk of producing a fetus inheriting no functional α-globin genes (ζ--/ζ--), leading to hemoglobin (Hb) Bart’s hydrops fetalis syndrome (BHFS). Expression of the embryonic α-globin genes (ζ-globin) is normally limited to the early stages of primitive erythropoiesis, and so when the ζ-globin genes are silenced, at ∼6 weeks of gestation, there should be no α-like globin chains to pair with the fetal γ-globin chains of Hb, which consequently form nonfunctional tetramers (γ4) known as Hb Bart’s. When deletions leave the ζ-globin gene intact, a low level of ζ-globin gene expression continues in definitive erythroid cells, producing small amounts of Hb Portland (ζ2γ2), a functional form of Hb that allows the fetus to survive up to the second or third trimester. Untreated, all affected individuals die at these stages of development. Prevention is therefore of paramount importance. With improvements in early diagnosis, intrauterine transfusion, and advanced perinatal care, there are now a small number of individuals with BHFS who have survived, with variable outcomes. A deeper understanding of the mechanism underlying the switch from ζ- to α-globin expression could enable persistence or reactivation of embryonic globin synthesis in definitive cells, thereby providing new therapeutic options for such patients.
Learning Objectives
Understand the concept of globin switching and the globin switches in gene expression that occur at the α- and β-globin loci
Understand the frequency of severe α-thalassemia, along with the causative genetic mutations and their outcomes
Understand the preventative screening, diagnosis, and management of severe α-thalassemia in utero
Appreciate the molecular mechanisms responsible for embryonic ζ-globin repression and how these may be manipulated to develop novel treatments for severe α-thalassemia
Background
In humans, the production of red blood cells occurs in 3 distinct waves.1 First, a cohort of cells originating in the yolk sac between days 17 and 21 is released into the circulation as the heart begins to beat at day ∼21. This cohort of primitive cells matures in a semisynchronous manner, and eventually, the cells enucleate. These primitive cells are no longer detectable after ∼12 weeks of gestation. On the basis of work in mice and human embryos, it is likely (although not yet definitely proven in humans) that a second transient wave of hematopoiesis, also originating in the yolk sac at day ∼23, migrates to the fetal liver and produces both erythroid and myeloid cells; in mice, these progenitors are referred to as EMPs.2,3 Erythroid cells derived from EMPs are considered to be early definitive cells. At day 28, cells arising from the splanchnopleura migrate to the floor of the dorsal aorta, where they undergo a transition to definitive hematopoietic stem cells that subsequently contribute to blood formation throughout life. By ∼70 days, all new red cells are derived from these definitive hematopoietic stem cells.
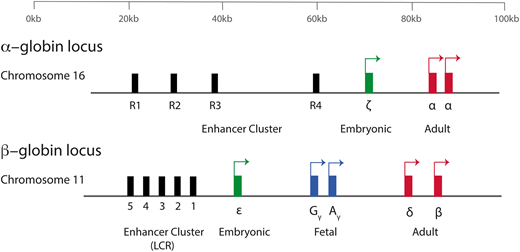
The production of hemoglobin (Hb) during development in humans is encoded by the α-like and β-like multigene clusters4 (Figure 1). The α-globin cluster is located on the short arm of chromosome 16 and includes the ζ-globin gene linked to 2 α-globin genes, arranged along the chromosome in the order in which they are expressed in development (5′-ζ-α2-α1-3′). Each haplotype is represented as ζ αα/, and therefore, the normal genotype is ζ αα/ζ αα. It has been previously shown that in humans, the embryonic ζ-globin gene is only expressed briefly in primitive erythroid cells between days ∼35 and 42 of gestation and that even in these cells, there is a rapid maturational switch from ζ- to α-globin expression (Figure 2).2 The ζ gene is normally silenced in all definitive erythroid cells.
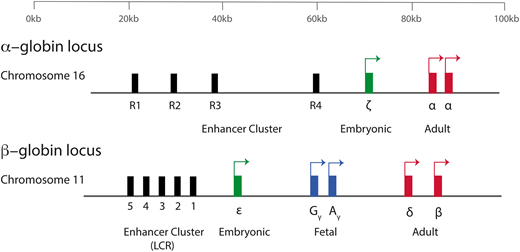
A simplified diagram of the human α-globin and β-globin loci. Embryonic globin genes are shown in green, fetal globin genes in blue, and adult globin genes in red. The α-globin genes are driven by 4 conserved enhancer elements (denoted R1-R4). The β-globin locus is driven by an enhancer cluster termed the locus control region (LCR).
A simplified diagram of the human α-globin and β-globin loci. Embryonic globin genes are shown in green, fetal globin genes in blue, and adult globin genes in red. The α-globin genes are driven by 4 conserved enhancer elements (denoted R1-R4). The β-globin locus is driven by an enhancer cluster termed the locus control region (LCR).
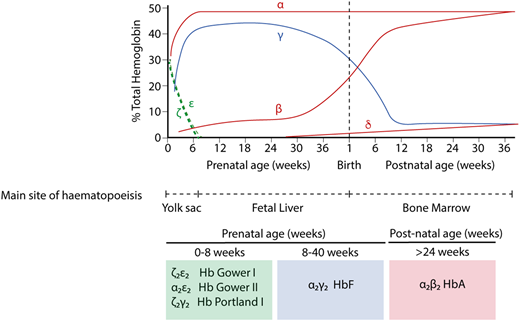
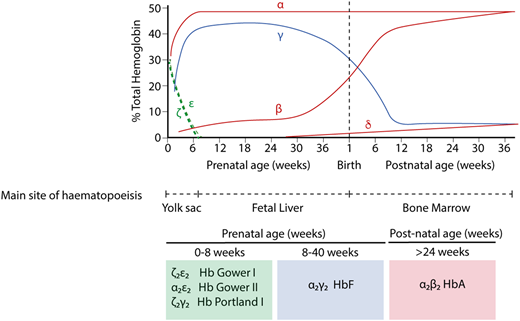
Hb switching at the human α- and β-globin loci. Embryonic globins are shown in green, fetal globins in blue, and adult globins in red. Primitive erythropoiesis, derived from the yolk sac, is characterized by expression of ζ-globin (from the α-globin locus) and ε-globin (from the β-globin locus). These are silenced at ∼8 weeks of gestation. α-globin then accounts for the entirety of the transcriptional output from the α-globin locus. At the β-globin locus, there is a switch to a fetal globin (γ-globin) during fetal life and then a second switch to the adult β-globin. The predominant type of Hb corresponding to each developmental stage is shown below.
Hb switching at the human α- and β-globin loci. Embryonic globins are shown in green, fetal globins in blue, and adult globins in red. Primitive erythropoiesis, derived from the yolk sac, is characterized by expression of ζ-globin (from the α-globin locus) and ε-globin (from the β-globin locus). These are silenced at ∼8 weeks of gestation. α-globin then accounts for the entirety of the transcriptional output from the α-globin locus. At the β-globin locus, there is a switch to a fetal globin (γ-globin) during fetal life and then a second switch to the adult β-globin. The predominant type of Hb corresponding to each developmental stage is shown below.
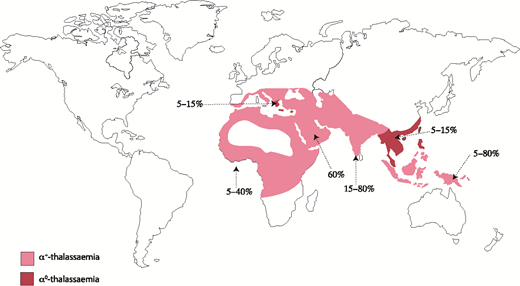
Naturally occurring mutations in the human α-globin cluster are found at high frequencies throughout all tropical and subtropical regions of the world, and it seems that such variants have been selected because carriers of α-thalassemia have a selective advantage in areas where Plasmodium falciparum malaria is endemic.5 α-thalassemia is most commonly caused by 1 of >50 deletions removing either 1 (ζ-α; referred to as α+-thalassemia) or both (ζ--; referred to as α0-thalassemia) α-globin genes.6 Some deletions remove all α-like globin genes (- --), and single point mutations may also cause nondeletional forms of α-thalassemia (ζ αTα).6 Αll known mutations are reviewed elsewhere and updated regularly.7 Α map denoting the distribution of mutations leading to α+-thalassemia and α0-thalassemia is shown in Figure 3.
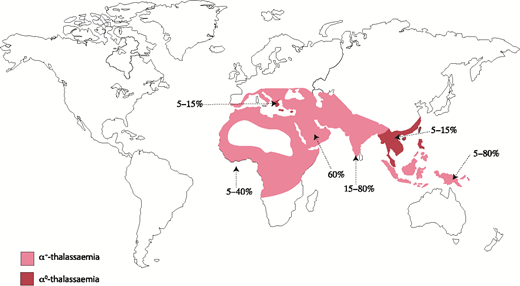
Prevalence and distribution of the mutations leading to α-thalassemia globally. Prevalence varies locally, hence the range of values.
Prevalence and distribution of the mutations leading to α-thalassemia globally. Prevalence varies locally, hence the range of values.
One particular deletion, ζ--SEA, occurs at high frequency (3% to 14%; Figure 4) throughout Southeast Asia, and homozygotes for this allele (ζ--SEA/ζ--SEA) have Hb Bart’s hydrops fetalis syndrome (BHFS), a lethal form of anemia.8,9 In some regions of Southeast Asia, as many as 1:200 neonates have this condition; it is thought that this balanced polymorphism results from a strong selective advantage of the ζ--SEA allele in such areas. Similar deletions affecting the α-globin cluster occur at much lower frequencies in other regions of the world, and so BHFS is rarely encountered in individuals other than those from Southeast Asia.
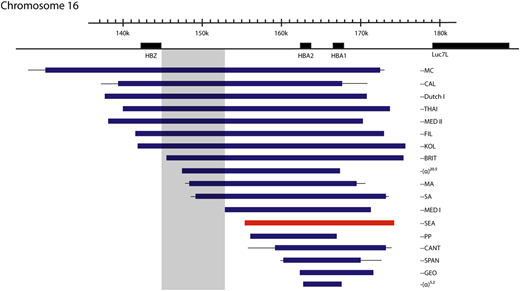
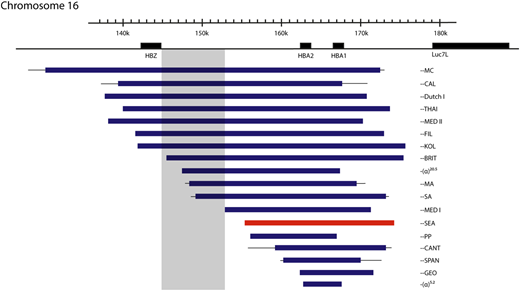
Mutations that cause deletional severe α-thalassemia. Lines indicate uncertainty in the breakpoints. The most common mutation, the ζ‐‐SEA mutation, shown in red, accounts for 80% of such mutations worldwide. Mutations encroaching upon the shaded area have no detectable ζ-globin in definitive erythropoiesis, despite the other promoters in the locus being deleted, suggesting a possible unidentified regulatory region.
Mutations that cause deletional severe α-thalassemia. Lines indicate uncertainty in the breakpoints. The most common mutation, the ζ‐‐SEA mutation, shown in red, accounts for 80% of such mutations worldwide. Mutations encroaching upon the shaded area have no detectable ζ-globin in definitive erythropoiesis, despite the other promoters in the locus being deleted, suggesting a possible unidentified regulatory region.
Infants with BHFS have the most severe deficiency in α-globin expression. Nonfunctional homotetramers of fetal γ-globin chains (γ4) make up most of the Hb in the erythrocytes of affected infants. They are profoundly anemic, with an average Hb of 64 g/L; embryonic Hb Portland (ζ2γ2), which is the only functional oxygen carrying Hb in these infants, accounts for ∼20% of this.10 The cardinal clinical features are those of a pale edematous infant with signs of cardiac failure and prolonged intrauterine anemia. Affected fetuses have pronounced hepatosplenomegaly, retardation in brain growth, and a wide range of developmental abnormalities, and there is gross enlargement of the placenta. Infants with BHFS almost always die either in utero (23-38 weeks) or shortly after birth. This condition also carries a serious risk, including mortality, to the mother of the affected fetus, with up to 50% developing severe preeclampsia, at least in part because of placentomegaly.11
Prevention of Hb BHFS and outcomes in long-term survivors
Given the high frequency of this genetic disease in people of Southeast Asian origin, the lethal degree of anemia, the associated developmental abnormalities, and the risks to the lives of pregnant mothers, prevention of this condition is of paramount importance. The most important diagnostic criteria to detect carriers of all types of thalassemia are microcytosis (mean corpuscular volume <80 fL) and/or hypochromia (mean corpuscular Hb <27 pg).8 When such indices are found in a pregnant woman, this should prompt a full diagnostic algorithm to determine if she carries thalassemia and if so which type.12
The partner of any carrier with α-thalassemia should be tested to determine if he too carries α-thalassemia and the risk of the fetus having BHFS established. Subsequent fetal diagnosis includes ultrasonography, chorionic villus sampling, or fetal blood sampling, as previously described.8 Successful screening programs are region and hospital dependent, with some centers reporting excellent compliance and almost complete BHFS prevention.13 Worldwide, however, BHFS babies continue to be born or only identified late in gestation, almost always resulting in perinatal death of the fetus and serious maternal complications.8
Although many affected fetuses have the expected features of BHFS, there may be considerable variability in the intrauterine clinical course. Some pregnancies, especially those resulting from large α-thalassemia deletions, including the embryonic ζ gene (- --) terminate unnoticed or early in gestation. Others continue throughout pregnancy and are stillborn or critically ill at birth. Some do not become hydropic and are born spontaneously.9,14 The environmental and genetic factors responsible for such diverse intrauterine courses of BHFS are currently unknown.
With improving methods of intrauterine diagnosis and neonatal care of critically ill newborn infants, there are increasing numbers of reports of affected fetuses being treated with intrauterine transfusion and neonates being treated by intensive care and transfusion. This was first reported in 1986,15 and a recent review identified and reported the outcome in 69 affected individuals.9 In most cases, such management resulted either because parents wished to continue with the pregnancy despite the diagnosis or because the condition was not diagnosed until the time of birth. To date, 39 of the 69 patients have survived beyond the age of 5 years, and 18 are now age >10 years; the oldest surviving patient is in her mid 30s. Surviving patients require lifelong blood transfusion and iron chelation, although at least 16 have undergone successful bone marrow transplantation.9,14 Developmental abnormalities are commonly seen in BHFS. It is likely that fetal hypoxia disturbs organogenesis and fetal development. Many anomalies have been reported, including microcephaly, hydrocephaly, hypoplasia of lungs, anomalous genitalia, and cardiac defects. Because anemia develops as soon as the ζ-α switch occurs, many abnormalities will occur before any of the current diagnostic tests can detect an affected fetus. From the small number of patients studied, even with optimal postnatal treatment, growth retardation and some degree of neurodevelopmental delay seem to be major adverse long-term outcomes.
These findings reemphasize the importance of successfully implementing a prenatal screening program and unbiased genetic counseling of the parents of an affected fetus.
Understanding how the embryonic ζ-globin gene is silenced in adult erythropoiesis
Studying developmental switches is of considerable scientific interest but also raises the possibility of reactivating expression of normal embryonic or fetal genes, which, if switched back on, could fully replace the activity of the missing or faulty adult genes underlying human diseases. Significant progress has been made in recent years in understanding the mechanisms and proteins involved in the silencing of ζ-globin expression, thereby opening up novel potential therapeutic avenues.
Cis-acting factors affecting ζ-globin expression
Expression of α-globin in adult erythroid cells is governed by 4 highly conserved cis-regulatory elements (enhancers). These regulatory sequences are termed multispecies conserved sequences R1 to R416 ; 3 (R1-R3) lie within introns of the upstream housekeeping NPRL3 gene. Naturally occurring mutations in combined with knockout mouse models for each of the enhancer elements indicate that R1 and R2 are the most important regulatory elements for α-globin transcription in adult erythroid cells.17-19
We have recently shown that the ζ-globin gene is dependent upon the same enhancer elements as α-globin during primitive erythropoiesis (currently under review), and the self-interacting erythroid domain is identical between primitive and definitive erythroid cells in mice. Somewhat surprisingly, the ζ-globin gene remains in contact with its enhancers in definitive erythroid cells (although at a lower frequency than in primitive cells), despite being silenced; this means the silencing mechanism is more complex than simple movement away from its active enhancers.
In humans with the ζ--SEA deletion on 1 chromosome, increased levels of ζ-globin are detected,20 and fetuses with BHFS have ∼20% Hb Portland (ζ2γ2).10 This suggests that in the absence of competition from the α-globin genes, interaction with the enhancers can partially overcome silencing of the ζ-globin gene. This is not the case, however, for all mutations causing deletion of both α-globin genes, with, for example, the larger ζ--BRITand ζ--SA mutations (Figure 4) having no detectable ζ-globin.21 This may mean that active regulatory regions necessary for ζ-globin expression in definitive erythroid cells reside within a 5.2-kb region present in the ζ--SEA deletion but absent in the ζ--BRITand ζ--SA chromosomes, although no cis-regulatory elements are identified in this region using open chromatin assays.
Candidate cis-acting candidate epigenetic silencing mechanisms for ζ-globin in definitive erythropoiesis include histone modifications (including histone deacetylation), DNA methylation, and recruitment of the polycomb repressive complex. The lack of a CpG island in the ζ-globin promoter suggests that DNA methylation is less likely to be important, and chromatin immunoprecipitation (ChIP) data show that H3K27me3 (the histone mark associated with PRC2 complex recruitment) is absent in definitive erythropoiesis, pointing against this as a silencing mechanism.22 Given that transcription factors implicated in embryonic silencing lead to histone deacetylation, it is probable that this plays an important role.
Trans-acting factors responsible for ζ-globin repression
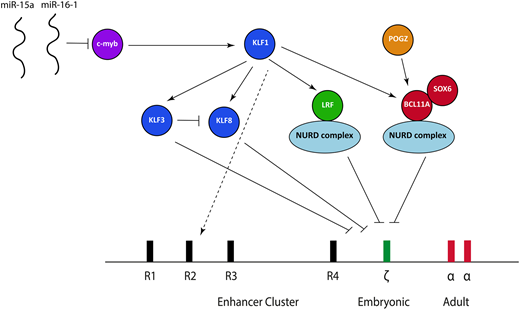
Over the last 5 years, some aspects of the transcriptional factor network responsible for embryonic ζ-globin globin gene repression have been elucidated (Figure 5), based predominantly on coincident work studying the fetal γ-globin to adult β-globin switch at the β-globin locus in both human cells and mice. It is now clear that the 2 main transcription factors responsible for repressing γ-globin in definitive erythroid cells, B-cell lymphoma/leukemia 11A (BCL11A) and leukemia/lymphoma-related factor (LRF), also play a role in ζ-globin repression.
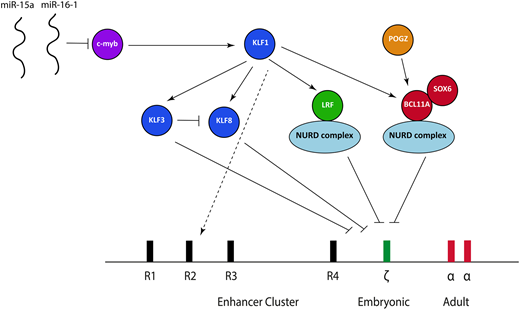
The transcriptional factor network currently known to be important in suppressing ζ-globin expression. miR, microRNA; NURD, nucleosome remodeling and deacetylation.
The transcriptional factor network currently known to be important in suppressing ζ-globin expression. miR, microRNA; NURD, nucleosome remodeling and deacetylation.
BCL11A is a multizinc finger transcription factor that was initially implicated in mediating the switch between γ-globin and β-globin at the β-globin locus based upon findings obtained from genome-wide association studies.23-25 Knockdown or knockout of BCL11A in human primary erythroid cells26 and animal models27,28 combined with the study of BCL11A-haploinsufficent individuals later confirmed that BCL11A is an integral γ-globin silencer.29 Aside from the effects at the β-globin locus, transcriptome data from an erythroid-specific BCL11A knockout mouse model show significant ζ-globin derepression,28 and we have confirmed these findings in both mouse and human cells. BCL11A ChIP sequencing in human cells also shows direct binding to the ζ-globin promoter in an immortalized human definitive erythroid cell line.30 The consensus binding site for BCL11A identified from recent ChIP sequencing studies,30,31 TGACCA, is present in 2 locations within the ζ-globin promoter, with 1 of these lying adjacent to a CCAAT box (TGACCAAT). Mutations within an identical sequence at the human γ-globin promoter within the β-globin locus lead to hereditary persistence of fetal Hb (HPFH), and disruption of that sequence using CRISPR/cas9 causes γ-globin derepression.30-32 It is therefore probable that BCL11A exerts its effects via direct binding at the ζ-globin promoter.
The zinc finger transcriptional repressor LRF (also known as Pokemon, osteoclast-derived ZF, FBI-1, and ZBTB7A) is a member of the BTB/POZ-ZF family; it is known to bind directly to DNA as a homodimer and leads to transcriptional silencing via chromatin modifications.33 Recent work points to a critical role for LRF in silencing ζ-globin (and its murine equivalent) in mouse and human cells.34 Analysis of a conditional LRF knockout mouse model35 showed LRF-deficient erythroblasts have marked upregulation of the ζ-globin gene with impaired erythroid differentiation34 ; these findings have also been confirmed in an immortalized erythroid human cell line where CRISPR/cas9 was used to knock out LRF.34 ChIP sequencing data shows direct binding to the ζ-globin promoter in both mouse and human cells.34
Exactly how BCL11A and LRF lead to ζ-globin gene repression remains an open question. The leading hypothesis is that this occurs via recruitment of the nucleosome remodeling and deacetylase (NURD) complex. The NURD complex is a highly conserved, multiprotein chromatin remodeling factor initially defined as a transcriptional repressor36,37 ; its proteins have both histone deacetylation (eg, HDAC1 and 2) and ATP-dependent chromatin remodeling activity (CHD4 and CHD3).38 Both BCL11A and LRF coassociate with individual members of the NURD complex,34,39 and knockdown of the CHD4 protein causes derepression of both the fetal and embryonic globin genes at the β-globin gene in human erythroid cells, strongly suggesting a role in developmental globin gene regulation.40 Despite LRF and BCL11A both coassociating with the NURD complex, they seem to act independently; our unpublished data suggest that knockout of both has a greater effect on ζ-globin than individual knockout. Interestingly, the NURD complex is capable of both activation and repression,41 implying a model more complex than simple recruitment of those proteins to the ζ-globin gene followed by silencing. Recent work, for example, has shown that components of the NURD complex are bound to the α-globin promoter in a definitive murine cell line, where it seems to act as a coactivator.42
A number of other transcription factors are also involved in ζ-globin gene repression, acting either together with BCL11A and LRF or upstream. SOX6, a member of the Sox transcription factor family characterized by the conserved high-mobility group domain,43 seems to interact physically as a protein complex with BCL11A, at least at the β-globin locus. A SOX6-null mutant mouse has been shown to express ζ-globin,44 and we have confirmed these findings in a definitive erythroid cell line. There is therefore clear evidence that SOX6 plays a role in embryonic globin gene repression, although the mechanism underlying this is unclear.
KLF1, the founding member of the 17-member family of KLF, is a master erythroid transcription factor defined by the presence of 3 similar C2H2 zinc fingers at the C terminus that comprise its DNA binding domain45 ; it is responsible for a wide activation repertoire, including genes involved in protein stabilizing, the heme biosynthetic pathway, the red cell membrane, and the cell cycle.45,46 A small number of individuals with a variety of KLF1 mutations and elevated ζ-globin expression have been described. These cases also have raised γ-globin expression, associated with chronic nonspherocytic hemolytic anemia.47,48 All of these cases have now been shown to harbor biallelic mutations in the gene encoding KLF1, causing deleterious effects in DNA binding, thus providing clear in vivo evidence that KLF1 plays an essential role in embryonic globin gene repression.
KLF1 activates both BCL11A49 and LRF,50 and it is therefore probable that at least some of the effects of KLF1 are mediated via these 2 transcription factors. KLF1 also activates 2 transcriptional repressors from the KLF family: KLF3 and KLF8, which bind MCS-R2 and the α-globin promoter and exert their effects via recruitment of the CtBP protein complex,51 which in turn causes epigenetic silencing via histone modifications.52 A study involving knockout of KLF3 and KLF8 individually and in combination in mouse models demonstrated that both seem to be involved in repressing the embryonic globin genes (including ζ-globin), with each partially compensating for the other when individually knocked out; KLF3 also inhibits KLF8 expression, suggesting a potential fine-tuning mechanism.53 Another option aside from upstream effects of BCL11A, LRF, KL3, and KLF8 is that KLF1 exerts additional effects by binding directly within the α-globin locus,54 although it does not directly bind the α-globin or ζ-globin promoters.50
c-myb, which is dispensable in primitive erythropoiesis but essential for definitive erythropoiesis, is a known trans-activator of KLF155 and has been implicated in silencing the embryonic and fetal genes at the β-globin locus.23 This implies that c-myb acts directly through KLF1 to influence BCL11A and LRF; such a model, however, would ignore the multitude of other transcriptional networks affected by c-myb, and there are probably other effects. Patients with trisomy 13 (Patau syndrome) have elevated fetal and embryonic globins.56 Chromosomal mapping of partial trisomy cases has implicated 2 miRs (miR-15a and miR–16-1) as causative; a combination of functional and genetic studies have demonstrated that these downregulate c-myb, thereby offering a plausible explanation as to how trisomy 13 leads to persistent fetal and embryonic globin gene expression.56
Finally, POGZ, a zinc finger binding transcription factor, has recently been shown to regulate both embryonic and fetal globins (including ζ-globin) in mouse and human cells.57 At least part of this effect is mediated via direct regulation of BCL11A, although it seems that it also represses the embryonic globin expression via other, as yet unidentified, pathways.
Somewhat surprisingly, of these trans-acting factors, only BCL11A, SOX6, and c-myb show an appropriate developmental pattern of expression, being absent in primitive erythropoiesis and highly expressed in definitive erythropoiesis.58 It may be that the other transcription factors undergo different posttranslational modifications or associate with different, unidentified binding partners in primitive and definitive erythroid cells. This observation points toward a model somewhat more complex than that currently conceived.
Strategies to reverse the embryonic globin switch in severe α-thalassemia
Although persistent ζ-globin expression to a level sufficient to salvage severe α-thalassemia has never been reported, transgene experiments in a mouse model indicate that continued expression of ζ-globin is sufficient to rescue a lethal α-thalassemia model,59 suggesting this is a viable therapeutic strategy. A potential concern is that Hb Portland 1 or 2 (ζ2γ2 or ζ2β2), which would be the predominant Hb produced when α-globin expression is absent in adults, both have higher oxygen affinities (evidenced by a lower P50 value) than conventional adult Hb.60 However, experience from patients with other high-affinity Hbs, some of which have lower P50 values than Hb Portland 2, suggests this is highly unlikely to have negative sequelae.61
There are 2 main options to derepress ζ-globin; pharmacological therapy or genome editing. Pharmacological targeting of the epigenetic mechanisms responsible for ζ-globin silencing is possible using small-molecular inhibitors; as a proof of principle, inhibition of histone deacetylation leads to ζ-globin expression in an in vitro primary erythroid cell culture system.62 Phenotypic screening of large drug libraries using primary erythroid cells and a sensitive readout for ζ-globin is currently being undertaken to identify compounds capable of activating ζ-globin expression, and this may allow development of new therapies.
Separately, it may be possible to use nuclease-based genome editing strategies to disrupt key regulatory elements or transcription factors in patient-derived CD34+ stem cells involved in ζ-globin silencing, followed by autologous transplantation of edited cells in infants salvaged by intrauterine transfusion.
Given that LRF and the KLF family of transcription factors play key roles in erythroid differentiation and lineage fate decisions, their targeting may have limited therapeutic potential. By contrast, knockout of BCL11A causes no discernible erythroid phenotype apart from its effects on globin expression.28 In addition, recent identification of an erythroid-specific BCL11A enhancer provides an attractive target for gene editing, causing erythroid knockdown of BCL11A while preserving normal B-cell function (in which BCL11A plays a vital role).63-65
An alternative genome editing approach would be targeting of key transcription factor binding sites in the ζ-globin gene promoter. The most attractive of these approaches would be to target the TGACCAAT BCL11A binding site, given that disruption of an identical sequence in the γ-globin gene causes HPFH and has been disrupted successfully using CRISPR/cas9 in human cells.30-32 We are currently exploring this strategy.
Future directions
There is a clear temporal difference between ζ-globin silencing (which occurs at ∼8 weeks gestation in humans) and γ-globin silencing (which starts at birth). It is therefore likely that in erythropoiesis, there are other, as yet unidentified, silencing and/or activating networks unique to embryonic globin gene expression. Many of the key discoveries underlying our understanding of the γ-globin to β-globin switch have resulted from analyzing large cohorts of patients with HPFH using genome-wide association studies.23-25,62 Similar large cohorts of patients with variable ζ-globin levels have not yet been studied, mainly because any persistent expression would almost certainly be at a low level and difficult to identify using standard Hb assays. However, studying patients with ζ--SEA on 1 allele and an intact locus on the other may circumvent this problem, given these patients already have some derepression of ζ-globin. We are in the process of recruiting and genotyping such a cohort. Complementary to this, the development of pooled CRISPR library screening66 should allow large-scale screening for both activating and repressive transcription factors.
An important and unanswered question is how much knockdown of BCL11A or disruption of its binding (or perturbation of another single transcription factor involved in ζ-globin silencing) would be required to upregulate ζ-globin to clinically beneficial levels when both α-globin genes are deleted. This situation is quite different to that found with strategies aiming to upregulate γ-globin in the β-globin hemoglobinopathies or thalassemias, when the adult β-globin gene promoters are usually intact. Because patients with BHFS already express 10% to ∼20% Hb Portland I ζ2γ2 in definitive erythroid cells,10 it is entirely plausible that removal of 1 repressive transcription factor alone may lead to sufficient levels of ζ-globin to ameliorate the severe α-thalassemia phenotype. This requires further exploration in vivo in animal models.
Clinically, advances in bone marrow transplantation and fetal medicine have led to the possibility of intrauterine bone marrow transplantation. There is currently a phase 1 clinical trial performing such transplantations using maternal (haploidentical) donors in fetuses affected by severe α-thalassemia between 18 and 25 weeks gestation (registered at www.clinicaltrials.gov as #NCT02986698). It is unknown at the moment whether the lack of conditioning and immunosuppression in these cases will permit adequate engraftment and allow these children to become transfusion independent. There are 3 previous reports on intrauterine bone marrow transplantation for severe α-thalassemia; none demonstrated successful engraftment or had positive outcomes.67 Considerable efforts are being made using preclinical mouse models to improve rates of engraftment in fetal transplantation. One successful strategy has involved depletion of endogenous hematopoietic stem cells using an antibody directed against c-kit (which is widely expressed on early hematopoietic cells).68 However, the mice used in this study were immunodeficient, and therefore, it remains unclear whether such an approach would translate to the treatment of hemoglobinopathies.
Acknowledgments
This work was supported by Medical Research Council Award 4050189188 (D.H.) and Wellcome Trust Clinical Training Fellowship 108785/Z/15/Z (A.J.K.).
Correspondence
Douglas R. Higgs, MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, University of Oxford, Oxford OX39DS, United Kingdom; e-mail: doug.higgs@imm.ox.ac.uk.