Abstract
Immunotherapies have been successfully developed for the treatment of B-cell acute lymphoblastic leukemia (B-ALL) with FDA approval of blinatumomab, inotuzumab, and tisagenlecleucel for relapsed or refractory patients. These agents target either CD19 or CD22, which are both expressed on the surface of the leukemic blasts in the majority of patients. The use of these agents has greatly transformed the landscape of available treatment, and it has provided curative therapy in some patients. As the field has matured, we are learning that for most patients, the currently available immunotherapies are not curative. Leukemic resistance to both CD19 and CD22 pressure has been described and is a major component of developed resistance to these therapies. Patients with B-ALL have developed CD19- or CD22-negative B-ALL, and in more rare cases, they have undergone lineage switch to acute myeloid leukemia. Current efforts are focusing on overcoming antigen escape, either by forced antigen expression or by dual-targeting therapies. A functional immune system is also required for maximal benefit of immunotherapy, particularly with chimeric antigen receptor (CAR) T-cell therapies. Data are now being produced that may allow for the prospective identification of patients whose immune deficits may be identified up front and predict failure. Preclinical work is focusing on additional engineering of CAR T cells to overcome these inherent immune deficits. Last, with improved knowledge of which patients are likely to benefit from immunotherapy as definitive treatment, those patients who are predicted to develop resistance may be prospectively recommended to undergo a consolidative hematopoietic cell transplant to lessen the recurrence risk.
Learning Objectives
Identify the major barriers to curative intent with current immunotherapy approaches for pediatric B-cell acute lymphoblastic leukemia (B-ALL)
Discuss newer immunotherapies that are currently under investigation to overcome barriers to treatment in pediatric B-ALL
Learn which patients may benefit from a consolidative transplant after immunotherapy for pediatric B-ALL
Clinical case
A 13-year-old girl was diagnosed with high-risk B-cell acute lymphoblastic leukemia (B-ALL), restricted to the marrow, at the age of 11 years. After up-front treatment, she presented with early combined marrow and central nervous system (CNS) relapse. After reinduction therapy, she continued to have persistent disease, with 19% residual blasts by flow cytometry in her marrow. Her leukemic blasts were noted to express CD10, CD19, partial CD22, and terminal deoxynucleotidyltransferase. Because of the chemorefractory nature of her leukemia, she received 1 month of continuous infusion blinatumomab at a dose of 5 μg/m2 × 7 days followed by 21 days at 15 μg/m2/d dosing. She experienced no toxicities but continued to have persistent CD19+ disease. She went on to receive CD19-directed chimeric antigen receptor (CAR) T-cell therapy at a dose of 1 × 106 CAR T cells per kilogram and achieved a complete remission (CR) that was minimal residual disease (MRD) negative 1 month after infusion. Despite this favorable response, she lost persistence of her CAR T cells and had recovery of normal B cells at 3 months after infusion. On the basis of her early B-cell recovery, her treating physician decided to pursue a hematopoietic cell transplant (HCT) with an unrelated donor. She is now at 1 year post-HCT and remains in remission.
Introduction
Over the past decade, immunotherapy advances for the treatment of B-ALL have been transformational. Both antibody- and cell-based therapies have enabled relapsed and refractory patients to obtain remission. Despite successes in achieving initial remissions, the number of patients ultimately cured is limited. This article presents mechanisms preventing cure from immunotherapy treatments and reviews strategies being investigated to improve remission durability. Failures of immunotherapy can be explained by intrinsic leukemic properties whereby blasts are resistant to immunotherapeutic pressures or the immune system’s inability to induce and/or sustain remission (Table 1; Figure 1).
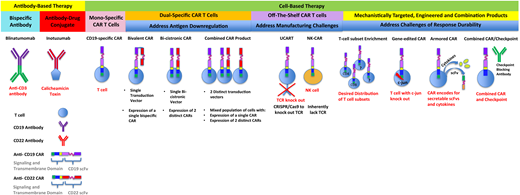
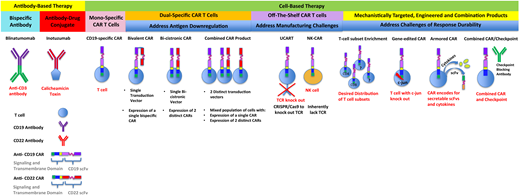
Antibody-based and cell-based immunotherapies that have been clinically translated or are under development. Included are next-generation strategies to address challenges that limit responses and durability of responses after treatment with monospecific chimeric antigen receptors.
Antibody-based and cell-based immunotherapies that have been clinically translated or are under development. Included are next-generation strategies to address challenges that limit responses and durability of responses after treatment with monospecific chimeric antigen receptors.
Innate leukemic resistance to targeted immunotherapy
Most available immunotherapy treatments for B-ALL target either CD19 or CD22. An advantage of both targets is expression early in B-cell development and continued expression through the mature B-cell stage, with resultant expression on the vast majority of B-ALL leukemic blasts.1,2 Three of these antigen-directed immunotherapies are currently FDA approved for use in B-ALL. Blinatumomab is a bispecific T-cell–engaging antibody that targets CD19 and CD3, allowing the T cells to be redirected to CD19 target cells.3,4 Inotuzumab ozogamicin (InO) is a CD22-specific drug–antibody conjugate with a calicheamicin payload.5 Tisagenlecleucel is a CD19-targeting CAR T-cell product.6
Antigen remodeling as a dominant pattern of relapse
CD19 loss as a mechanism of relapse was reported early after blinatumomab and CAR T cells were introduced to the clinic, and it has emerged as one of the most significant barriers to curative outcomes. The impact of CD19 loss is highlighted in the pediatric CAR T-cell experience, where the global phase II study (NCT02435849) identified CD19 antigen loss in 15 of 16 evaluable relapse samples.6 In addition, a mechanistic study analyzing relapsed samples derived from 2 phase II CD19 CAR T-cell trials (NCT02435849 and NCT02228096) in pediatric patients identified leukemia to be CD19 negative in 12 of 17 relapsed samples.7 CD19 loss in adults treated with CAR T cells has been reported with relatively decreased frequency, with CD19-negative relapse accounting for 30% of post-CAR T-cell relapses. CD19 loss after blinatumomab, however, is reported to be a less frequent phenomenon. In the pediatric phase I/II study of blinatumomab (NCT01471782), the CR rate approached 40%, with 71% of these patients relapsing within 6 months, 22% (4 of 18) of these relapses being CD19 negative.8 The first multi-institutional publication describing outcomes after blinatumomab failure in adults with B-cell malignancies reported only 8% of relapses (5 of 61) to be CD19 negative.9
Mechanistic studies of antigen downregulation
The first comprehensive analysis of CD19 antigen escape after CD19-specific therapy identified splice variants of CD19 that restrained surface expression of the epitope recognized by CD19-specific CARs.10 Follow-up work demonstrated that splice variants and in-frame mutations result in misfolded proteins that are retained in the endoplasmic reticulum and unable to travel to the cell surface.11 Complementary work analyzing relapsed samples derived from CD19 CAR T-cell trials in pediatric patients identified that every CD19-negative sample under study (N = 12) had a minimum of one frameshift mutation that was predicted to result in a truncated CD19 protein lacking capacity to anchor CD19 to the membrane.7 A single report identified CD19 isoforms, later accounting for resistance, to be expressed at initial diagnosis before CAR T-cell treatment.12 Aside from this single report, the majority of studies have been unable to identify mutations in CD19 driving antigen loss in pretreatment samples. Work studying mechanisms of CD19 downregulation after blinatumomab similarly describe unchanged cell phenotypes before and after blinatumomab, with the exception of CD19 alterations, supportive of isolated mutational events driving relapse.13
Lineage switch as a mechanism of resistance
In addition to antigen loss, cases of lineage switch have occurred after CD19 targeting. Initial reports of lineage switch were in patients with KMT2A (lysine methyltransferase 2A) rearrangements and occurred shortly after CAR T-cell infusion.14 Subsequent reports described cases of lineage switch after blinatumomab,15 as well as in broader groups of patients, including B-ALL lacking KMT2A translocations, and temporally occurring several years after CD19 targeting.16,17 For patients with KMT2A rearrangements, partner chromosomes may differentially impact the rate of lineage switch.18 In vivo mouse models were developed that recapitulate lineage switch and propose lineage reprogramming to be driven by antigenic pressure.19 With improved mechanistic understanding of lineage switch, it may become possible to identify patients at highest risk up front and adjust treatment strategies to avoid prolonged targeted pressure or consolidate targeted therapy with HCT.
Role of successive CD19-targeted immune therapies in driving antigen-negative relapse
The first report of CD19-negative ALL occurred in a patient who first received blinatumomab, followed by CAR T cells.20,21 A recent, large retrospective review of 150 patients raised concern that prior treatment with blinatumomab increased the risk of CD19-negative disease.22 Interestingly, the level of expression of CD19 on the leukemic blasts before treatment was not predictive of antigen escape. On the contrary, a retrospective review of 24 adult patients treated with CD19 CAR T cells demonstrated that prior blinatumomab did not influence the risk of CD19-negative relapse.23 Future systematic study will provide a more definitive analysis of the impact of prior blinatumomab use on CAR T-cell outcomes.
CD22 as an alternative B-ALL target and resistance to CD22 targeting
CD22 targeting has shown great promise, both with CD22-directed CAR T-cell therapy and with InO. After CD22-directed CAR T-cell therapy (NCT02315612), remission rates are comparable to those with CD19 CAR T cells, with 73% obtaining a CR at therapeutic dosing.24 However, the rate of relapse after CD22-directed CAR T-cell therapy is much higher, with only 3 of the 12 responding patients remaining in CR, with a median remission duration of 6 months. The expression of CD22 is more variable in B-ALL than expression of CD19, driving a different mechanism of leukemic escape.1 Decreased CD22 site density or downmodulation is commonly seen after CD22 targeting as compared with the antigen loss after CD19 targeting.
In a retrospective review of 51 pediatric patients who received InO, 67% achieved CR, and only 3 had evidence of CD22 downmodulation.25 In an adult study of InO, similar CR rates were achieved in patients despite varying levels of CD22 expression, including patients with CD22 blast expression as low as 50% to 69%.26 This sensitivity to downmodulation may be less apparent with InO, possibly because of a bystander effect from the calicheamicin toxin. Certain subgroups of B-ALL, such as Philadelphia chromosome (Ph) positive and KMT2A, have less CD22 expression, and adult studies revealed that these subgroups were less responsive to InO.26 This finding was not validated in the pediatric retrospective analysis.25
Investigations introducing compounds to upregulate and maintain expression of CD22 are underway. Bryostatin 1, a macrocyclic lactone compound, has been shown to specifically upregulate the CD22 antigen and has improved efficacy of CD22-targeted therapies in preclinical models.27,28 By forced upregulation of CD22, it may be possible to increase remission durability with CD22-targeting therapies.
Development of dual-targeting approaches and respective challenges
Because of the large number of recurrences related to antigen escape, several groups are now investigating dual-targeting approaches with CAR T cells. The greatest clinical experience to date combines CD19 and CD22. Tandem CAR constructs, also referred to as bispecific or bivalent constructs, contain an extracellular binding domain that is capable of dual-antigen recognition fused to a single intracellular signaling moiety. A tandem CAR capable of recognizing CD19 and CD22 was developed by Qin et al29 and is undergoing clinical evaluation for both pediatric and adult B-cell malignancies (NCT03241940 and NCT03233854, respectively), with preliminary signal of efficacy reported at the 2018 American Society of Hematology annual meeting.30 Tandem CAR constructs may pose technical challenges in enabling continued antigen recognition and activity against both targets. An alternative approach uses a product whereby individual T cells express 2 distinct CARs. This can be achieved in 2 ways. The first is through dual transduction with 2 vectors, resulting in a heterogeneous population of CAR T cells with both single- and dual-CAR expression. This approach is being investigated in the PLAT-05 trial (NCT03330691) with preliminary CR rates of 83% in CAR-naive patients.31 Dual-expressing CAR T cells can also be produced using a single bicistronic vector that allows for all transduced T cells to express both CAR constructs. Great Ormond Street Hospital has taken this approach, with initial results of its phase I study (NCT03289455) demonstrating a 100% MRD-negative remission rate in 6 patients treated at 3 × 106 CAR T cells per kilogram and ongoing molecular CR and B-cell aplasia in 4 of 6 patients at last follow-up.32
All these trials carry the risk of simultaneous downregulation of CD19 and CD22, generating aberrant clones stripped of targetable surface antigens, restraining further salvage with alternative available therapies such as blinatumomab or InO. New targets are being developed, such as the CAR targeting the thymic stromal lymphopoietin receptor antigen frequently overexpressed in Ph-like B-ALL33 and CD123,34 which will broaden possibilities for multiplex targeting.
Immune system resistance
Immune-regulatory signals and exhaustion challenge the durability of immunotherapy responses
Immune-regulatory signals can impair the efficacy of antibody-based therapy. Regulatory T cells (Tregs) have been shown to constrain outcomes in patients receiving blinatumomab. Blinatumomab-activated Tregs mediate immune suppression through interleukin-10 (IL-10) production and inhibit T-cell proliferation and cytotoxicity. Enumeration of Tregs in patients who received blinatumomab demonstrated that higher levels of peripheral blood Tregs correlate with nonresponders.35 Checkpoint expression can also limit T-cell responses in antibody-based therapies. Preclinical data combining antibody-based therapies with checkpoint blockade showed enhanced T-cell proliferation and cytotoxicity.36
CAR T-cell persistence has been shown to be a desirable outcome in B-ALL because it correlates with sustained clinical remission and is protective against CD19+ recurrence.6,37 Although initial work demonstrated that the costimulatory domain of the CAR construct impacts the durability of the in vivo persistence, with 4-1BB costimulation protective against exhaustion as compared with CD28,38,39 additional work is ongoing to evaluate other biological features of the product and cell engineering that can impact persistence.
Determination of optimal T-cell–intrinsic properties
T-cell–intrinsic properties can drive persistence and exhaustion. T-cell subsets, including the naive, central memory, stem cell memory, and effector memory compartments, carry different functional properties and predispositions toward achieving long-term immune memory. Although there is not yet consensus regarding the optimal T-cell component for adoptive therapy, preclinical studies have demonstrated that cells derived from less differentiated parental T-cell subsets are favorable for adoptive transfer.40 Combinations of varying subsets from CD4 and CD8 compartments can confer enhanced efficacy over using CD4 or CD8 subsets alone.41 Recent work noted that starting T-cell phenotypes in nonresponding patients have higher levels of LAG-3 (lymphocyte activation gene 3) and less tumor necrosis factor-α production.37 This same study showed that products high in tumor necrosis factor-α production and low TIM-3 (T-cell immunoglobulin and mucin protein 3) expression provide more durable engraftment.37 Further efforts are required to study and implement CAR T-cell therapy comprised of a defined, tailored combination of optimized T-cell subsets and to identify modifiable predictors of CAR persistence.
Novel engineering strategies to address T-cell exhaustion
Systematic CAR studies have demonstrated that small variances in CAR design impact biological function. Epigenetic studies employing an assay for transposase-accessible chromatin using sequencing to evaluate the landscape of tonically active, exhausted CAR T cells identify the AP1 (activator protein 1) family of transcription factors as important mediators of exhaustion. Specifically, skewing the balance of AP1 factors toward overexpression of c-Jun can reduce expression of inhibitory molecules and confer enhanced antitumor effect.42 Efforts to specifically tailor CAR engineering to ameliorate exhaustion are ongoing.
Conventionally, CARs are introduced into T cells through virally mediated transduction that yields random CAR integration. Work targeting CAR integration to the site of the T-cell receptor α constant (TRAC) using CRISPR/Cas9 technology demonstrated uniform CAR expression and enhanced potency. CAR placement under endogenous regulatory elements appears to preserve T-cell–naive and central memory phenotype markers. Furthermore, TRAC-CAR T cells are protected from accelerated exhaustion and yield improved antitumor activity in animal models.43
An alternative approach under preclinical study combines CAR T cells with checkpoint blockade through the development of CAR T cells that secrete checkpoint-blocking single-chain variable fragments. In vivo these “armored CARs” demonstrate similar or enhanced efficacy as compared with conventional approaches combining checkpoint blockade and CAR T-cell therapy. This strategy confers the possible advantage of decreased systemic toxicity because checkpoint delivery is local, concentrating effect to the tumor site.44
Enhancing efficacy and persistence through combinatorial approaches
Early-phase clinical trials combining checkpoint blockade and blinatumomab in adult and pediatric relapsed/refractory B-cell malignancies are ongoing (NCT02879695, NCT03605589) and preliminarily demonstrate feasibility and manageable toxicities.45 Similar efforts are underway to combine CAR T cells with checkpoint blockade. A single-center clinical trial (NCT02374333, NCT02906371) combined CD19 CAR T cells with either pembrolizumab or nivolumab in patients with heavily pretreated relapsed B-cell malignancies. Checkpoint inhibitors were administered a minimum of 14 days after CAR T cells in order not to overlap with peak CAR toxicities. Remarkably, 3 of 6 patients who received checkpoint blockade because of recovery of physiologic B cells were able to reestablish B-cell aplasia after checkpoint blockade. Patients with bulky extramedullary disease that persisted through CAR T cells or relapsed after CAR T cells achieved objective responses, with 2 of 4 patients achieving complete disease resolution.46
Immune-mediated rejection of CAR T cells
Immunologic rejection is another obstacle challenging CAR T-cell persistence. The majority of CD19 CARs integrated murine-derived single-chain variable fragments, introducing the possibility of human antimurine immune rejection.47 To address this, fully human or humanized binders have been developed to prevent transgene rejection and allow repeated dosing. Clinical remissions have been reported using humanized CAR constructs after failed murine-based CARs (NCT02374333).48
Immune privileged sites
To achieve antitumor effect, it is imperative that immune-based therapies traffic to sites of disease. Blinatumomab and InO have little ability to cross the blood–brain barrier. Increasing relapses with extramedullary disease are being reported after these therapies. A retrospective study in patients receiving blinatumomab identified a history of prior or active extramedullary disease as a predictor of lower CR rates. Furthermore, 40% of relapses occurred with extramedullary disease.49 CAR T cells can effectively cross the blood–brain barrier. Although early studies excluded CNS disease because of the concerns of neurotoxicity, it has subsequently been shown that CAR T cells can effectively treat CNS disease without impacting neurotoxicity.50,51 Data studying CAR T-cell trafficking to the CNS demonstrated that 98% of patients treated with CD19-specific CAR T cells had detectable CAR T cells in their cerebrospinal fluid at day 28 after treatment. This report described that CAR T-cell persistence in the cerebrospinal fluid correlated with persistence in the blood and bone marrow. The ability of CAR T cells to penetrate and effectively treat CNS disease is a benefit of CAR T cells over antibody-based therapies.50
Manufacturing challenges
In order to obtain clinical benefit from immunotherapy, a therapeutic product must exist. Antibody-based therapies are available “off the shelf” as soon as they are required for use. Autologous CAR T-cell products require several steps before delivery to the patient, which can introduce barriers in delivering the intended therapy.
Technical challenges with apheresis
Regardless of manufacturing strategies, product development is contingent on the ability to tolerate apheresis and obtain adequate lymphocytes for manufacturing. Work surveying the yield of apheresis in patients (N = 99) undergoing harvest for the purpose of CAR T-cell therapy demonstrated that despite heavy pretreatment and a broad absolute lymphocyte count (ALC) range at the time of collection (142-6944 cells/μL), all patients met the targeted mononuclear cell count of 1 × 109 cells. Notably, a subset of 22 patients with lower ALC counts <500 were successfully harvested, supporting the fact that adequate lymphocytes can be obtained even from heavily pretreated patients with low ALCs.52 Despite these encouraging data, for subgroups of patients, successful manufacturing cannot be achieved. Initial manufacturing failure rates were predicted to be as high as 25% because of poor lymphocyte quality.53 With the use of up-front cell selection, which depletes monocytes and other potential inhibitory cells, as well as the use of homeostatic cytokines such as IL-7, IL-15, and IL-21, cell expansion can be improved with up to 100% manufacturing success.53-55
Manufacturing complication yielding CAR expression on leukemic blasts
One concerning report demonstrated aberrant CD19–CAR transduction of a single leukemic B cell during manufacturing, which resulted in relapsed disease with a CAR-expressing leukemic clone 9 months after infusion. CAR expression on the leukemic cell bound and masked the CD19 epitope on the cell surface, making this clone unrecognizable to CAR and resistant to CAR-mediated cytotoxicity.56
Off-the-shelf products
Despite advances in manufacturing, vulnerable patients continue to exist for whom autologous products are not an option. These patients include very small infants who do not meet size requirements to undergo apheresis, patients who have received intensive lymphodepleting chemotherapy and cannot meet required washout periods, and those with recurrence early after HCT before immune reconstitution. For these groups of patients, work is ongoing to develop off-the-shelf universal cell products.
Off-the-shelf CAR T-cell products
Technical challenges that exist for allogeneic CAR T-cell products include the risk of inducing graft-versus-host disease (GVHD) through the native T-cell receptor (TCR) of the CAR T cells and rejection of the CAR T cells by the patient. The most mature clinical data using allogeneic CAR T cells have been generated from Great Ormond Street Hospital using UCART19 (NCT02746952 and NCT02808442).57,58 To permit crossing of allogeneic barriers, this off-the-shelf product has its TCR knocked out using transcription activator–like effector nucleases directed at the TRAC locus to prevent GVHD. CD52 is also knocked out to render CAR T cells resistant to alemtuzumab. Patients receive alemtuzumab before CAR T-cell infusion to induce profound lymphodepletion and prevent rejection of the third-party T cells. Results to date have shown modest success with this strategy, with CR rates of 67% and further increases in CR to 82% in those patients who received alemtuzumab. Owing to the short engraftment of the CAR T cells, for long-term remission, patients must be quickly bridged to HCT.
CAR-expressing natural killer cells
An alternative approach to generating off-the-shelf CAR cellular products is to genetically engineer natural killer (NK) cells to express CARs.59 NK cells are part of the innate immune system and broadly recognize foreign cells in an HLA-unrestricted manner. In contrast to T cells, NK cells uniquely can achieve cytotoxicity without the risk of GVHD because they lack TCRs. NK cell lines, such as NK-92, have been generated and tested clinically with a tolerable safety profile, and they have been under investigation as a source of off-the-shelf allogeneic effector cells for adoptive therapy. These cells are short-lived effectors and do not have the potential for persistence and long-term immune memory. One potential benefit of CAR-NK cells is the possibility of continued nonspecific cytotoxicity in the face of antigen downregulation through the NK cell–mediated innate immune response. Until recently, the majority of CAR-NK studies have been preclinical; however, clinical trials are currently being initiated.59-61 In addition to using NK-92 cells, researchers have begun to manufacture NK cells derived from cord blood with NK cells engineered not only to express the CAR but also to secrete IL-15 and express the suicide safety switch, inducible caspase 9. A phase I/II clinical trial of this NK cell product with a CD19-specific CAR is currently underway (NCT03056339).62
Determination of the role of HCT after immunotherapy
Antibody-based therapies
The durability of response using antibody-based therapy continues to be evaluated as data accumulates from early studies. A global pediatric phase I/II study (NCT01471782) evaluating blinatumomab in relapsed/refractory ALL demonstrated a CR rate of 39% after 1 or 2 cycles. Long-term follow-up data showed survival rates of only 20% at 24 months. Although there was heterogeneity in who went to transplant, yielding a small sample size for study, overall survival did not differ between transplanted and nontransplanted patients. A trend toward prolonged survival was noted in patients achieving MRD-negative CR.63 Long-term phase II follow-up data in adults investigated for the role of consolidative stem cell transplant after blinatumomab suggested that HCT after blinatumomab increased survival in patients <35 years old who received blinatumomab for MRD. The survival benefit is lost in patients >35 years of age, suggesting that the role for HCT may be greatest in patients treated with blinatumomab with MRD who can tolerate HCT.64 For InO, an adult retrospective review analyzed subjects who received InO and then proceeded to HCT. HCT-naive subjects who proceeded directly to HCT had the best 2-year survival probability of 51%, as opposed to those subjects who had a prior history of HCT or those whose remission from InO was lost before HCT.65 A prospective study is currently planned to better define the benefit of HCT after InO.
Cell-based therapies
Some patients treated with CD19-specific CAR T cells without subsequent antileukemic therapy have maintained long-term remission for >5 years. For most patients, however, currently available CAR T cells are not definitive therapy. Identifying those patients who are at risk of not sustaining a durable remission is critical in determining who should proceed with HCT after CAR T cells. This has been an area of substantial controversy without consensus. Complicating the matter is the mixed results of pediatric and adult studies in determining whether HCT after CAR T cells provides a survival advantage. The experience of adults treated with a CD19-CD28 CAR showed no advantage for those who proceeded with HCT.66 A pediatric retrospective review (N = 50) showed a statistically significant benefit of a consolidative HCT on leukemia-free survival in HCT-naive patients and those who lost CAR T-cell persistence before 2 months after infusion.67 A limitation of this study included small patient numbers and its retrospective nature.
Conclusions
As seen in the present clinical case, immunotherapy can allow otherwise refractory patients to obtain deep remission, but often resistance can be seen. In the present clinical case, blinatumomab was unable to induce remission. However, resistance to blinatumomab did not confer resistance to CAR T cells. Likewise, repeated targeting of CD19 through different modalities can enable clinical efficacy, although more data are needed to truly understand if cumulative single-antigen targeting increases the risk of antigen loss. Further CD19 targeting was chosen in this patient rather than switching to CD22 targeting, owing to the heterogeneous expression of CD22. Consideration could have been given to InO, which does not require uniform CD22 expression to induce remission. After CAR T cells, the patient was at significant risk of recurrence because of the short in vivo persistence. This was addressed by pursuing consolidative stem cell transplant, especially in the context of the patient being HCT naive.
With the current repertoire of available immunotherapies for B-ALL, some patients can continue to be salvaged and put into a state of chronic disease. With increasing sophistication of CAR T-cell design, including dual-targeting approaches as well as methods to enhance persistence, it is likely that more patients will be cured with CAR T cells. The role of antibody therapy as definitive treatment is less clear. We anticipate that further clinical experience will inform its curative potential. We also expect the field to advance in identifying patient-specific variables with predictive value to guide the optimal choice of immunotherapy for each patient.
Correspondence
Rebecca Gardner, Seattle Children’s Hospital, 4800 Sand Point Way NE, Seattle, WA 98105; e-mail: rebecca.gardner@seattlechildrens.org.