Abstract
Emerging methods to detect tumor-derived DNA in the blood plasma of patients with lymphomas—so-called “circulating tumor DNA” (ctDNA)—have the potential to change the way in which lymphoma is diagnosed and managed in the clinic. The possible applications for ctDNA are numerous, including mutation genotyping, response monitoring, and detection of minimal residual disease during a time of radiographic remission. This article discusses the methodology for detecting ctDNA in aggressive B-cell lymphomas, including digital polymerase chain reaction, targeted sequencing of immunoglobulin receptors, and targeted next-generation sequencing. The advantages of each of these methods are also compared, with a focus on promising clinical applications. These include identification of molecular subtypes (eg, cell-of-origin and double-hit lymphomas) from pretreatment plasma, molecular response prediction after an initial course of therapy, and early detection of relapsing disease prior to clinical relapse. Finally, this article discusses the challenges in implementing ctDNA assays in the clinic today, including possible solutions to these challenges.
Learning Objectives
Distinguish among the multiple methodologies for measuring ctDNA in lymphomas, including the advantages of each method
Understand when ctDNA has potential clinical utility, including in tumor genotyping and MRD monitoring for relapse and response detection
Patient case
A 42-year-old female presented to a local urgent care center with new onset of left lower quadrant abdominal pain and fullness. A physical examination revealed a firm palpable mass rising to the level of the umbilicus. Magnetic resonance imaging of the pelvis revealed a 13 × 9 × 20-cm mass adjacent to the left ovary and an obstructed right ureter. She underwent a laparoscopic biopsy, which demonstrated a large B-cell lymphoma with expression of CD10, CD19, and CD20, resulting in a diagnosis of diffuse large B-cell lymphoma (DLBCL), germinal center subtype by the Hans algorithm. Fluorescence in situ hybridization (FISH) for MYC, BCL2, and BCL6 was performed and was negative. A positron emission tomography (PET)/computed tomography (CT) scan demonstrated fluorodeoxyglucose-avid disease in the left pelvic mass with a maximum standardized uptake value of 20.5 and no disease above the diaphragm. Her International Prognostic Index (IPI) was 1 (elevated lactate dehydrogenase; low risk category). DNA from the initial tumor biopsy and pretreatment plasma were sequenced via Cancer Personalized Profiling by Deep Sequencing (CAPP-Seq), demonstrating coding alterations in TP53, CREBBP, and STAT6 shared between the 2 samples. The patient was treated with an initial cycle of RCHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) therapy; circulating tumor DNA (ctDNA) was still detectable directly prior to cycle 2 of therapy, with a 1.8-log reduction in tumor DNA compared with pretreatment levels. She completed 6 cycles of RCHOP therapy, achieving a complete response by PET/CT scan at the end of therapy. Three months after completion of RCHOP, she developed pain and fullness in her left inguinal region. A repeat PET/CT scan demonstrated new inguinal and external iliac lymphadenopathy. Subsequent CT-guided biopsy confirmed relapsed DLBCL.
Introduction
Technologies to detect malignancies noninvasively through the blood—so-called “liquid biopsies”—have emerged as important diagnostic tools in the last decade.1,2 Many hematologic neoplasms, such as leukemias, have circulating malignant cells that enable minimal residual disease (MRD) detection from peripheral blood mononuclear cells (PBMCs).3-5 Although a few lymphoma subtypes, such as cutaneous T-cell lymphomas and mantle cell lymphoma (MCL), can have high burdens of circulating tumor cells, some of the most common lymphoma subtypes, including DLBCL and classical Hodgkin lymphoma (cHL), do not circulate in high levels. Therefore, detection of disease in circulating plasma-derived cell-free DNA (cfDNA), rather than in circulating cells, is an attractive option for molecular evaluation in lymphomas. All people have a small amount of circulating cfDNA in their plasma, typically between 5 and 10 ng of DNA per milliliter, which is equivalent to ∼1000 to 2000 cells’ worth of DNA content.6 However, in patients with a cancer such as lymphoma, a small fraction of this total cfDNA will contain tumor-specific somatic alterations or circulating tumor DNA (ctDNA). Because the fraction of tumor-derived DNA can be <1% of total cfDNA in many cases, methods with low background noise are required to accurately detect and quantify ctDNA in patient samples.
Methods for ctDNA detection in lymphoma
Although many low-error profile methods for detection of ctDNA have been described, these largely fall into 2 major groups: polymerase chain reaction (PCR)-based assays and next-generation sequencing (NGS)-based assays (Table 1). PCR-based methods with low background, such as digital PCR (dPCR), have found clinical utility in diseases with recurrent truncal mutations, such as KRAS-mutated colon cancer and EGFR-mutated lung cancer.7-9 By dividing DNA molecules into individual partitions (ie, droplets) prior to amplification, dPCR allows for improved sensitivity and quantitation compared with traditional quantitative PCR. After PCR amplification, individual droplets can be enumerated for the presence of a specific mutation (ie, KRAS G12D or EGFR L858R) using fluorescent probes. This methodology is ideally suited for assessing “hotspot” mutations. As a result of significant interpatient heterogeneity in mature lymphoid neoplasms, few alterations are recurrent with sufficient frequency to prove useful for disease detection via PCR-based approaches. Previous efforts have demonstrated detection of MYD88 L265P in primary central nervous system lymphoma and lymphoplasmacytic lymphoma10,11 or XPO1 E571K in cHL via dPCR,12 serving as a proof-of-principle; however, only a fraction of all lymphoma patients carry these alterations.
In contrast, NGS-based approaches have the potential to capture somatic alterations in a larger fraction of patients as a result of their ability to assess multiple genetic regions, including the immunoglobulin loci (ie, IGH, IGL, and IGK), which are clonally rearranged in most B-cell lymphomas. An initial approach focused on high-throughput amplicon sequencing of the immunoglobulin loci (IgHTS; or the clonoSEQ assay) has been applied in multiple lymphoma subtypes (Table 1).13,14 Using consensus primers to amplify only rearranged VDJ sequences, followed by sequencing library construction, IgHTS allows sensitive detection of lymphoma-specific clonotypes from cellular DNA or cfDNA. In a cohort of patients with DLBCL, IgHTS demonstrated superiority for disease detection from ctDNA compared with PBMCs. However, dPCR and IgHTS are limited by their reliance on only a single genetic locus. Indeed, because each milliliter of plasma contains only ∼1000 to 2000 cell equivalents of DNA, the nominal sensitivity for disease detection is limited to ∼1 part in 2000 when assessing a single mutation from a single milliliter of plasma, or ∼1 in 10 000 from a typical 5-mL plasma sample.
Two alternatives exist to increase the limit-of-detection: increasing the amount of input material or assessing multiple genetic alterations or mutations. When assessing ctDNA levels using multiple genomic alterations, disease detection does not require seeing every mutation, but only some of the tumor-defining alterations, thereby allowing detection at time points of low disease burden. Targeted ultradeep NGS platforms to detect multiple mutations per individual case have emerged, including hybrid capture–based approaches (ie, CAPP-Seq6,15-18 ) and amplicon-based approaches (ie, Lymphopanel19 ) (Table 1). Hybrid capture– and amplicon-based approaches leverage an ultra-high depth of sequencing by focusing on a relatively small portion of the genome. By sequencing thousands of unique molecules at each position while leveraging PCR-induced error suppression, these methods enable sensitive detection of residual disease from plasma DNA. Monitoring ctDNA levels via targeted sequencing first requires identification of tumor-specific alterations, typically via sequencing of tumor DNA or pretreatment plasma DNA, along with matched germline DNA to remove constitutional variants. This results in a list of tumor-specific alterations that can then be interrogated in future samples to measure ctDNA levels. By targeting multiple recurrently mutated regions of the genome, including regions affected by single nucleotide variants (SNVs), insertions/deletions, genetic rearrangements, and copy number variations (CNVs), these approaches enable biological insights into tumor biology, as well as ultrasensitive disease detection. Compared with IgHTS, an initial approach with CAPP-Seq identified >100 independent mutations to enable tumor monitoring, thereby improving disease detection from 74% to 100% in pretreatment ctDNA samples from 23 DLBCL patients.16 Importantly, targeted-sequencing approaches focus on only a small fraction of the ∼3 billion bases in the human genome, sequenced to very high depth. Although this is useful for disease detection, alterations throughout the remainder of the genome will necessarily be missed. Alternative approaches for ctDNA detection from genome-wide assessment of CNVs are emerging,20,21 although the limit of detection for such assays is currently suboptimal.
Clinical utility of ctDNA detection and monitoring in lymphoma
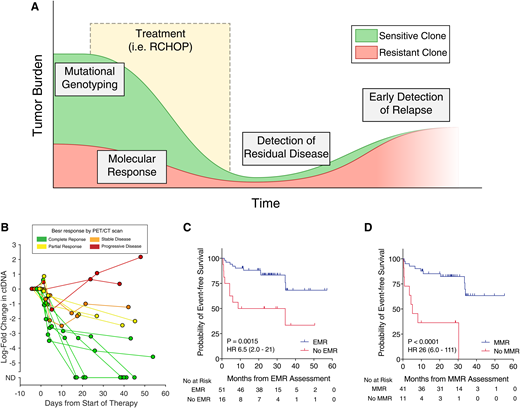
In the clinic, ctDNA has many potential roles for understanding the prognosis of patients with lymphomas. Previous studies have demonstrated the utility of ctDNA for genotyping lymphomas prior to therapy, detection of molecular response, and early detection of recurrence and resistance (Figure 1A). Here, I will examine the data supporting each of these use cases in turn.
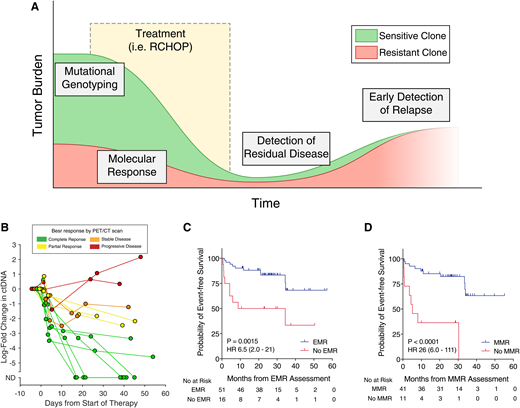
Clinical uses of ctDNA in lymphoma. (A) An idealized schematic of the course of a patient through initial diagnosis, treatment, and relapse with lymphoma. ctDNA assessment has potential applications throughout the disease course, including mutational genotyping prior to treatment, molecular response assessment at early time points during treatment, detection of residual disease at the end of therapy, and early detection of relapsed disease prior to eventual clinical relapse. (B) The dynamics of ctDNA levels in 14 patients with dense serial sampling are shown as a spider plot. Levels of ctDNA are normalized to pretreatment levels, whereas each line is colored according to the patient’s eventual best response by PET/CT scan. (C) Kaplan-Meier estimates of EFS for patients achieving or not achieving a 2-log decrease in ctDNA after 1 cycle of therapy (ie, EMR). (D) Kaplan-Meier estimates of EFS for patients achieving or not achieving a 2.5-log decrease in ctDNA after 2 cycles of therapy (ie, MMR). Data in (B-D) are reproduced from Kurtz et al18 with permission. HR, hazard ratio.
Clinical uses of ctDNA in lymphoma. (A) An idealized schematic of the course of a patient through initial diagnosis, treatment, and relapse with lymphoma. ctDNA assessment has potential applications throughout the disease course, including mutational genotyping prior to treatment, molecular response assessment at early time points during treatment, detection of residual disease at the end of therapy, and early detection of relapsed disease prior to eventual clinical relapse. (B) The dynamics of ctDNA levels in 14 patients with dense serial sampling are shown as a spider plot. Levels of ctDNA are normalized to pretreatment levels, whereas each line is colored according to the patient’s eventual best response by PET/CT scan. (C) Kaplan-Meier estimates of EFS for patients achieving or not achieving a 2-log decrease in ctDNA after 1 cycle of therapy (ie, EMR). (D) Kaplan-Meier estimates of EFS for patients achieving or not achieving a 2.5-log decrease in ctDNA after 2 cycles of therapy (ie, MMR). Data in (B-D) are reproduced from Kurtz et al18 with permission. HR, hazard ratio.
Noninvasive pretreatment plasma genotyping
A number of clinically important molecular features have been described in aggressive B-cell lymphomas, including translocation events in MYC, BCL2, and BCL622 and recurrent mutations in individual genes, such as MYD88, CD79B, EZH2, CREBBP, and TP53.23-25 Typically, these alterations are identified by assessing diagnostic material from tumor biopsies by FISH for translocations or targeted NGS for SNVs. However, identification of tumor mutations from tissue is potentially limited as a result of possible spatial heterogeneity (ie, genetic differences that can occur between multiple distinct tumor sites) and the possibility of limited tumor material. Indeed, because diagnostic samples are now commonly obtained from fine needle aspirations and core needle biopsies, limited material for diagnostic testing is frequently encountered in the clinic. Detection of somatic mutations directly from a blood draw via ctDNA is an attractive alternative; it is capable of sampling multiple tumor regions simultaneously without significant sampling limitations. Multiple studies in DLBCL have demonstrated robust concordance between somatic SNVs identified from sequencing from paired tumor and plasma samples; for example, in an initial study of 45 patients with DLBCL with paired pretreatment tumor and plasma samples, >90% of mutations in driver genes seen in the tumor sample were also seen in the plasma.16 Similarly, plasma ctDNA sequencing in 86 DLBCL patients revealed 78% concordance for detection of FISH-confirmed rearrangements in MYC, BCL2, and BCL6, including 82% (9/11) concordance in the detection of double-hit genotypes.26 Initial studies also support the ability to detect clinical meaningful CNV events, including amplifications in PD-L1, in diverse lymphoma subtypes.21 Although the robust performance of noninvasive plasma genotyping has primarily been shown in patients with DLBCL, initial studies suggest that these findings are broadly applicable to other lymphomas, including in >100 patients with cHL27 and 14 patients with peripheral T-cell lymphomas.28
Furthermore, somatic mutations in specific genes have previously been shown to occur preferentially in specific cell-of-origin (COO) subtypes; for example, MYD88 L265P and CD79B mutations preferentially occur in the activated B-cell subtype of DLBCL, whereas BCL2 translocations and CREBBP mutations preferentially occur in the germinal center B-cell subtype. Profiling of ctDNA mutations for identification of COO subtype has been demonstrated, with 77% concordance with tissue immunohistochemistry COO classification.16,29 In the above clinical case, ctDNA profiling by CAPP-Seq revealed mutations in CREBBP and STAT6, which are enriched in the germinal center B-cell subtype of DLBCL, consistent with the COO classification of her tumor using immunohistochemistry.
Quantification of ctDNA and molecular response assessment
One powerful aspect of ctDNA is the ability to quantitatively assess levels serially over time. Assessments of disease burden, such as total metabolic tumor volume measured by fluorodeoxyglucose PET/CT scan, have demonstrated prognostic significance in patients with lymphomas, including in DLBCL.30 In DLBCL, quantitative levels of ctDNA prior to therapy are significantly correlated with total metabolic tumor volume, as well as with clinical risk factors like the IPI, suggesting a potential role as a prognostic factor.18 Indeed, in a study using a training and validation framework across 217 patients from 6 international centers, our group demonstrated that pretreatment ctDNA level is a predictor of outcomes for patients with previously untreated DLBCL; patients with low ctDNA levels had superior event-free survival (EFS) compared with patients with high levels.
Although pretreatment levels of ctDNA are prognostic for outcomes in DLBCL, circulating biomarkers in other diseases, such as chronic myelogenous leukemia and chronic lymphocytic leukemia, suggest that detection of an initial response to therapy can be highly prognostic.3,4 As such, multiple independent studies have attempted to identify a “molecular response” in ctDNA-based MRD to predict eventual outcomes. An initial approach using IgHTS in serum samples from 126 patients with previously untreated DLBCL suggested that the presence of ctDNA after 2 cycles of dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) therapy identified a group with inferior time to disease progression.14 Building on this finding, our group attempted to identify prognostic thresholds to measure molecular response in DLBCL, as early as after 1 cycle of standard RCHOP therapy, using the more sensitive CAPP-Seq methodology (Figure 1B).18 Using DLBCL patients from 6 centers in a training and validation framework, our group demonstrated superior EFS for patients achieving ≥2-log reduction in ctDNA compared with pretreatment levels after a single cycle of first-line anthracycline-based therapy (ie, RCHOP or dose-adjusted EPOCH-R). This same threshold was also prognostic for EFS after salvage therapy. We termed this 2-log reduction in ctDNA after 1 cycle of therapy an “early molecular response” (EMR; Figure 1C). Similarly, patients achieving a 2.5-log decrease in ctDNA after 2 cycles of therapy also had superior EFS; we termed this threshold a “major molecular response” (MMR; Figure 1D). It is also notable that EMR and MMR were more strongly prognostic of outcomes than pretreatment ctDNA levels; although patients with high vs low ctDNA had differences in EFS with a hazard ratio of 2.4 to 2.6, the hazard ratios for EMR and MMR were 6.5 to 10 and 11 to 26, respectively. Importantly, in multivariate models controlling for the IPI and interim PET/CT results, molecular response remained prognostic for EFS and overall survival. This independent prognostic value is essential as ctDNA moves forward in clinical paradigms.
Although the preponderance of the evidence for molecular response measurement from ctDNA exists in DLBCL, similar concepts have been applied to other lymphomas. Using CAPP-Seq, Spina and colleagues reported a similar 2-log threshold in ctDNA as being predictive of progression-free survival in patients with advanced-stage cHL receiving ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) (n = 24).27 Interim and end-of-therapy ctDNA levels by IgHTS in MCL patients undergoing first-line therapy (n = 53) have also been shown to be prognostic for outcomes.31 In the above case, the patient did have a decrease in ctDNA level after her first cycle of therapy, but her 1.8-log reduction in levels did not meet the criteria for an EMR, predicting possible poor outcome. Indeed, 3 months after completing 6 cycles of RCHOP therapy and achieving a complete response, the patient had multiple new hypermetabolic lymph nodes in her left pelvis on PET/CT scan, with a biopsy confirming relapsed disease.
Early detection of clinical relapse
Although many aggressive lymphomas are treated with curative intent, a significant fraction of patients will experience clinical relapse after achieving a first remission. As such, surveillance strategies ranging from frequent clinical examinations to radiographic imaging32 have been used to detect relapsed disease at the earliest possible time point. Thus, detection of molecular relapse via ctDNA-based MRD has been explored as a method for early detection of relapsed disease. Similar to noninvasive genotyping and molecular response assessment, the preponderance of evidence exists from DLBCL. Two independent studies utilizing IgHTS for disease detection after initial remission demonstrated that ctDNA became detectable with a median lead time of 3 to 3.5 months before clinical evidence of disease.13,14 More sensitive approaches offer the possibility of a longer lead time prior to clinically overt disease. Assessment of plasma DNA with CAPP-Seq in DLBCL patients leading up to clinical relapse demonstrated an average lead time of 6 months, outperforming IgHTS in 9 patients for whom paired samples were available.16 Earlier detection of relapsed disease via CAPP-Seq or other targeted-sequencing approaches also has the potential to inform on disease biology and the mechanisms of resistance to therapy. Exemplifying this, during treatment with the Bruton’s tyrosine kinase inhibitor ibrutinib, ctDNA analysis can reveal emergent BTK C481S mutations that drive resistance to this therapy.16,33 Tracking mechanisms of therapy resistance at relapse via ctDNA has also been demonstrated in MCL patients receiving ibrutinib and venetoclax.34 Beyond DLBCL, studies have demonstrated earlier detection of relapsed disease via ctDNA in diverse lymphomas, including using IgHTS in MCL35 and T-cell receptor sequencing in peripheral T-cell lymphomas.36
Challenges of translating ctDNA in the clinic
As described above, multiple independent studies have demonstrated the utility of ctDNA assessment for risk-stratifying lymphoma patients throughout a disease course. The fact that these findings are consistent across multiple groups using various methods for detecting ctDNA suggests that this biomarker is robust to preanalytical considerations with potential for translation into clinical practice. However, a number of logistic hurdles remain before real-time ctDNA assessment becomes accessible in clinical trials or routine clinical care. Previous studies were largely performed on archival samples collected and frozen as part of clinical studies; this has allowed sequencing to be performed in a retrospective manner. Prospective translation requires real-time sample processing and reporting, with turnaround times of ideally no more than 7 to 10 days required to make clinical decisions on the results of a test. Although various targeted NGS assays for ctDNA are available from multiple diagnostic companies, these panels largely focus on mutations found in solid tumors, rather than lymphomas or other hematologic cancers. For disease monitoring, IgHTS (ie, ClonoSEQ) from plasma or cellular samples is available for clinical use; however, the remainder of the methods described above have yet to be translated into the clinic for routine use in lymphomas.
Although versions of these assays are on their way to clinical laboratories, discrepancies in methodology will become more apparent when trying to compare between tests. Efforts to harmonize and standardize ctDNA quantification will be necessary, as they were with other biomarkers, such as PET/CT scans.37,38 The use of reference standards assessed on each platform, along with transparent reporting of methodologies and technical limits of detection, will be essential. Furthermore, although ctDNA has been shown to have independent prognostic value in multivariate models,18 it is not yet clear how to integrate this marker into risk-stratification frameworks with established biomarkers, including IPI, COO, and interim PET/CT scans; this is particularly true for serial ctDNA assessment over time. To best use ctDNA, including its unique ability for repeated measurements over time, new risk-stratification methods will be needed.
Finally, and perhaps most significantly, questions remain regarding how to act on ctDNA assessments. Although pretreatment and dynamic risk assessments are capable of identifying patients at high risk for treatment failure, how to improve outcomes for these patients remains unclear. Previous studies utilizing PET/CT scans or the IPI for risk-adapted approaches have largely failed to improve outcomes,39-41 potentially as a result of the imperfect sensitivity and specificity of these biomarkers. Therefore, randomized prospective trials exploring novel therapies for high-risk patients not achieving molecular response will be needed to demonstrate superior outcomes and ultimately lead ctDNA assessment into routine clinical care. Similarly, assessment of a “de-escalation approach” in low-risk patients achieving a favorable molecular response—similar to interim PET/CT negativity in cHL42 —is warranted. Finally, further studies will be required to establish the clinical utility of liquid biopsies in other lymphoma subtypes, including low-grade lymphomas in which clinical management strategies are distinct from more aggressive histologies. Therefore, defining the role of liquid biopsy in the clinical management of diverse lymphoma subtypes is an ongoing area of research; however, with the tools in hand to perform ctDNA assays, this area is ripe for prospective clinical translation today and in the near future.
Acknowledgments
This work was supported by the National Cancer Institute (K08-CA241076), Damon Runyon Cancer Research Foundation (PST #09-16), and the Conquer Cancer Foundation of the American Society of Clinical Oncology (Young Investigator Award).
Correspondence
David M. Kurtz, Division of Oncology, Department of Medicine, Stanford University, 1291 Welch Rd, SIM 1 Lokey Bldg, Rm G2115, Stanford, CA 94305; e-mail: dkurtz@stanford.edu.