

Abstract
The aggressive peripheral T-cell lymphomas (PTCLs) are a heterogenous group of uncommon lymphomas of mature T lymphocytes dominated by 3 subtypes: systemic anaplastic large-cell lymphoma, both anaplastic lymphoma kinase positive and negative; nodal PTCL with T-follicular helper phenotype; and PTCL, not otherwise specified. Although the accurate diagnosis of T-cell lymphoma and the subtyping of these lymphomas may be challenging, there is growing evidence that knowledge of the subtype of disease can aid in prognostication and in the selection of optimal treatments, in both the front-line and the relapsed or refractory setting. This report focuses on the 3 most common subtypes of aggressive PTCL, to learn how current knowledge may dictate choices of therapy and consultative referrals and inform rational targets and correlative studies in the development of future clinical trials. Finally, I note that clinical-pathologic correlation, especially in cases of T-cell lymphomas that may present with an extranodal component, is essential in the accurate diagnosis and subsequent treatment of our patients.
Learning Objectives
Understand the pressing need for accurate diagnosis and subtyping of peripheral T-cell lymphomas
Use the subtype of peripheral T-cell lymphoma to choose optimal upfront and subsequent therapies in an evidence-based manner
Much like their more common B-cell counterparts, T-cell lymphomas (TCLs) are a heterogenous group that comprise both indolent and more aggressive entities. In the 2016 SEER database, peripheral TCLs (PTCLs), those that originate from mature T lymphocytes, represent 5% of all non-Hodgkin lymphomas diagnosed in the United States.1 The most common of these are mycosis fungoides/Sezary syndrome, representing a generally more indolent subtype of PTCL and 3 generally more aggressive subtypes of PTCL that are the focus of this report: PTCL-not otherwise specified (PTCL-NOS); nodal PTCL with T-follicular helper (TFH) phenotype; and anaplastic large-cell lymphoma (ALCL). The aggressive PTCLs comprise not only these 3 entities but an additional 9 lymphomatous entities, all of which may be treated with multiagent regimens designed with curative intent2-4 (Table 1). Although the outcome of aggressive TCLs is far more dismal than that of their B-cell counterparts and indolent TCLs,1 subtyping of PTCLs has been increasingly used to help select optimum therapy and improve outcomes (Table 2).
Clinical case
The patient was a 36-year-old male smoker who noted right axillary lymphadenopathy. He was diagnosed with classic Hodgkin lymphoma by means of a node biopsy and was to have outpatient follow-up. Two months later, he presented to another institution with dysphagia and respiratory difficulty requiring intubation. A left cervical node biopsy was performed and was thought to represent classic Hodgkin lymphoma; the patient received doxorubicin, vinblastine, and dacarbazine. Computed tomographic scanning showed widespread adenopathy above and below the diaphragm. Initial laboratory test results were remarkable for elevated lactate dehydrogenase. A week later he had not improved, and a second-opinion pathology consultation was obtained. At that time, the diagnosis was changed to anaplastic large cell lymphoma (ALCL), anaplastic lymphoma kinase negative (ACK−). Morphologic and immunophenotypic evaluation of the cervical lymph node revealed an extensive sinusoidal infiltrate of large pleomorphic cells, with occasional crescentic hallmarks cells that stained positively for CD45, CD2, CD43, MUM1, c-Myc, CD30, CD15, EMA, and granzyme B and demonstrated a markedly increased proliferation rate (90%). Fluorescence in situ hybridization for DUSP22 and TP63 were negative. The patient received an additional dose of doxorubicin, cyclophosphamide, and prednisone (CHP) with growth factors and experienced rapid improvement with extubation. He received brentuximab vedotin (BV) and CHP (BV+CHP) for an additional 6 cycles and achieved a complete remission followed by high-dose chemotherapy with peripheral stem cell rescue. At this writing, he had been in complete remission for 1 year.
The perils of TCL diagnosis and classification
The diagnosis and classification of TCLs continue to present a challenge to both the diagnostic pathologist and the clinician, because the entities are uncommon; there is a lack of reliable markers of clonality; specific genetic alterations have not been identified for most entities; and there is a need, especially in lymphomas that involve extranodal sites, to integrate clinical features with the available pathologic information.8 In 2002, in a landmark study, the Non-Hodgkin’s Classification Project found that PTCL could be diagnosed reliably by an experienced hematopathologist but that immunophenotyping was absolutely necessary. 4 Indeed, a recent prospective study that included both academic and community practices showed that the diagnostic workup for PTCL in the United States continues to vary widely and often lacks important phenotypic information to fully characterize the lymphoma.9 The North American PTCL Study Group then undertook a project, suggesting that the use of a clearly defined algorithm as an approach to tissue diagnosis may render a diagnostic accuracy of greater than 90% in T-natural killer (T-NK) lymphomas10 (Figure 1). Consideration of the necessity for accurate diagnosis to inform prognosis and treatment has no doubt led to the current recommendation of the National Comprehensive Cancer Center Network (NCCN) that a “review of all slides with at least one paraffin block representative of the tumor should be done by a hematopathologist with expertise in the diagnosis of PTCL.”11 The NCCN also proposed as essential a tentative panel of immunophenotypic tests; however, the performance of these have not been prospectively evaluated.11
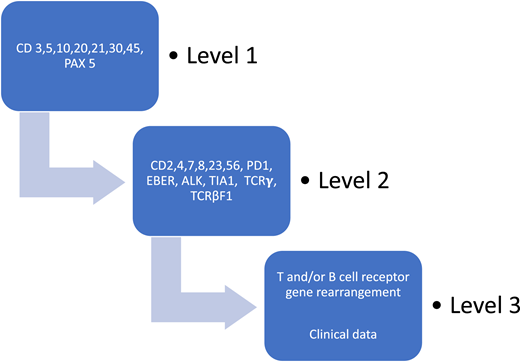
Algorithmic approach to diagnosis of PTCL after evaluation of basic demographics. Schematic of the case review algorithm. Adapted from the North American PTCL Study Group Project.10
Algorithmic approach to diagnosis of PTCL after evaluation of basic demographics. Schematic of the case review algorithm. Adapted from the North American PTCL Study Group Project.10
ALCL: defining optimal induction therapy and beyond
Systemic ALCL is perhaps the best example of a subtype of TCL where a diagnostic antigen, CD 30, can also function as an effective target for treatment. In ALCL, all tumor cells are strongly positive for CD30. CD30 is most strongly present at the cell membrane and in the Golgi region, although diffuse cytoplasmic positivity is also common. This diagnostic feature of ALCL has been exploited in the search for new treatments of ALCL in the form of an antibody-drug conjugate, BV, a drug consisting of a chimeric anti-CD30 monoclonal antibody linked to monomethyl auristatin E, a disruptor of microtubules.
First studied in the relapsed or refractory setting in a phase 2 trial, BV was found to have an 86% response rate with 57% complete responses (CRs) and a duration of response of 12.6 months12
After the publication of that study, the ECHELON-2 trial, a 452-patient global, randomized, double-blind, double dummy, phase 3 study comparing BV+CHP with CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) in the upfront treatment of patients diagnosed with PTCL, in which immunohistochemistry showed expression of CD30 in at least 10% of malignant cells, was published.13 In that trial, patients were stratified according to the International Prognostic Index (IPI) and histologic subtype. By design, the study was enriched for patients diagnosed with ALCL. Seventy percent of the patients with intent to treat had ALCL, with 22% of patients having anaplastic lymphoma kinase (ALK)–positive ALCL with ≥2 IPI. The patients received either 6 or 8 cycles of chemotherapy, predetermined by the investigator at each site; 19% of the patients received more than 6 cycles. With blinded independent review, the patients treated with BV+CHP had a statistically significant improvement in median progression-free survival (PFS), from 20.8 to 48.2 months, compared with those who received CHOP. Treatment with BV+CHP also reduced the risk of death by 34%, compared with CHOP. The study was not powered to perform subtype analysis. ECHELON-2 is the first randomized study of PTCL to show an overall survival benefit of a novel therapy combined with a standard backbone with no increase in toxicity.13
A post hoc exploratory analysis of the data looked at the effect of consolidative high-dose therapy with stem cell rescue (autologous bone marrow transplantation [ABMT]) in those patients with ALK− systemic ALCL and those with histologic results showing other than systemic ALCL. In the investigational arm, 67% of patients with ALK− systemic ALCL attained CR, with 36% of those undergoing ABMT. Fifty-nine percent of patients with histologic diagnosis other than systemic ALCL also attained CR, and 29% of those underwent ABMT. Patients treated with ABMT tended to be younger (median age, 50) and from non-Asian countries. Although the sample size was small, PFS estimates favored the use of ABMT.14
In the relapsed or refractory setting, the presence of a t(2;5) translocation involving the ALK and the nucleophosmin genes, which result in the expression of a novel fusion protein and overexpression of ALK in ALK+ ALCL, led to the study and effective use of crizotinib.15
The use of consolidative stem-cell transplant in the front-line treatment of PTCLs
Phase 216,17 and 3 studies14 have suggested higher rates of PFS in patients with PTCL who receive consolidative stem-cell therapy in the first CR, and this procedure has become standard practice at many institutions for patients with high-risk disease or histologic findings (Table 3). In most of these instances, the expected 5-year overall survival is significantly <50%, with the exception of late-stage breast implant–associated ALCL, which may be as low as 60%.18-21 In a prospective US cohort registry of patients with newly diagnosed PTCL, the Comprehensive Oncology Measures for PTCL Treatment (COMPLETE),23 of the 119 patients with nodal PTCL (ALK− ALCL, angioimmunoblastic TCL [AITL), and PTCL-NOS), who achieved complete remission after induction therapy, 36 patients underwent ABMT and 83 did not. Among the patients who underwent ABMT, a significantly higher number had advanced disease and high IPI, although there were no significant differences in demographics. With a median follow-up of 2.8 years, the median overall survival was not reached in the group receiving ABMT, whereas the group that did not undergo ABMT had a median overall survival of 57.6 months. Although this difference did not reach statistical significance (P = .06), in a multivariate analysis, autologous stem cell transplantation was independently associated with improved survival. The role of upfront ABMT as consolidation of first complete remission in the absence of validation in a prospective, randomized trial continues to be controversial.
AITL and other nodal lymphomas of TFH cell origin: selection of treatment in the relapsed or refractory setting and of rational targets for investigation
Since the recognition of TFH cells as a unique subset of T-helper (Th) cells with a characteristic phenotype, a subset of PTCLs has been noted to have a TFH phenotype. The most well studied of these is AITL. In addition, up to 40% of cases of PTCL-NOS24 have been found to share some of the clinical and pathologic features, the TFH phenotype, and some of the characteristic mutations of AITL. For this reason, in the 2016 revision of the World Health Organization classification of mature T neoplasms (an umbrella term), nodal lymphomas of TFH-cell origin, was introduced. For this designation, the malignant cell should express at least 2 or 3 TFH-related antigens including PD1, CD10, BCL6, CXCL 13, ICOS, SAP, and CCCR5. Recurrent genetic abnormalities associated with the TFH phenotype include the TET2, IDH2, DNMT3A, RHOA, and CD28 mutations, as well as gene fusions such as ITK-SYK or CTLA4-CD28.2,3
The recognition of this unique phenotype is advantageous in the classification of PTCL, enabling more specific diagnosis of some cases that previously would have been designated as PTCL-NOS, but identifying the function of the TFH cell in providing T-cell support to B lymphocytes may inform some of the clinical features of these conditions, such as the autoimmune phenomena and serologies, polyclonal hypergammaglobulinemia, and associated B-cell lymphomas.
Although the TFH phenotype has not yet been used to determine an induction therapy that would be most efficacious in these lymphomas, it has informed the selection of newer single agents that can be used for relapsed or refractory disease. Earlier uses of immunomodulatory drugs in treating AITL, such as cyclosporin and others, may serve as examples. Some of the elements of the TFH phenotype and its associated genetic mutations are, at this time, also serving as targets for novel agents in the clinical trial arena or may do so in the future.
Mutations of IDH2, TET2, DNMT3A, and RHOA, frequently seen in AITL as well as in PTCLs, particularly those with a TFH phenotype, involve genes that encode epigenetic modifiers. Histone deacetylase inhibitors (HDACis) are agents that act in part to increase acetylation of the histones associated with DNA, effecting their condensation within the nucleus of the cell. It is this modification of histones by acetylation that plays a key role in epigenetic regulation of gene expression. Both romidepsin, a class 1 selective HDAC, and belinostat, a class 1 to 4 HDACi, have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of relapsed and refractory aggressive PTCLs. Notably, the registration trials of these drugs showed that patients with AITL can have sustained responses with these agents, compared with other FDA-approved agents (Table 4).25-27
In AITL and in some cases of PTCL-NOS, CD30 overexpression has been noted in both the malignant T-cell compartment and in the B immunoblasts found in the microenvironment.35 These B immunoblasts may be positive or negative for Epstein Barr virus–encoded small RNA. This feature of AITL qualified subjects with a diagnosis of AITL for enrollment in the ECHELON-2 trial.13 Although 54 subjects with the diagnosis of AITL were randomized in the trial, they represented only 12% of the study population and cannot be analyzed separately. Treatment with BV+CHP represents a reasonable front-line therapy for patients with AITL, in which ≥10% of the malignant cells express CD30. In relapsed or refractory disease, a planned subset analysis of a study in which BV was administered to patients with CD30+ PTCLs included 13 subjects with AITL who were found to have an overall response rate of 54% with 38% CR.28
In the clinical trial arena, multiple agents are already available that could target the specific antigens in a TFH phenotype, such as ICOS (MEDI-570 in NCT02520791), IDH2 (AG-221), CXCL13, and IL21. The finding of Epstein Barr virus–positive B cells in 80% to 95% of cases of AILT3 would also commend trials with antiviral and HDACi combinations (www.clinicaltrials.gov, #NCT03397706). CD30 expression could also support the investigation of anti-CD30 chimeric antigen receptor T cells (#NCT04008394).
PTCL-NOS: future directions
Finally, PTCL-NOS is the heterogenous form of nodal and extranodal PTCL that does not correspond to any of the other specifically defined PTCL entities. Within this group, gene expression and microRNA profiling studies have delineated 2 biologic and prognostic subgroups: those with an increase in GATA-3 and those with an increase in TBX-21 expression.2,3,22,36 Some cases within the latter group may show cytotoxic differentiation. Each group also has been found to have specific genetic mutations. The genetic abnormalities in the GATA-3 group are more complex and include loss or mutation of tumor-suppressor genes targeting the p53 and Pi3 kinase pathways and gains in STAT-3 and myc.37 GATA-3 expression in PTCL-NOS is associated with an especially poor PFS and overall survival.36,38 The TBX-21 group is enriched for genetic mutations of genes regulating DNA methylation.38 TBX-21 and GATA-3 are transcription factors that regulate gene expression profiles in Th cells directing the Th cell into Th1- and Th2-cell–differentiating pathways, respectively. Immunohistochemical markers have been used in place of gene expression profiling and may have prognostic and therapeutic significance. Such observations provide biologic rationale for investigation of novel therapies for PTCLs, such as Pi3 kinase inhibitors, hypomethylating agents, and JAK/STAT pathway inhibitors, and may in future help us direct these therapies to the patients who may benefit most.
First appearances deceive many: the aggressive PTCL imposters
As the case history reported herein demonstrates, the accurate diagnosis of PTCLs and subtyping of these lymphomas is crucial to both prognostication and to enabling clinicians to make rational treatment decisions to benefit patients. Making these decisions may at times require collaboration with an experienced hematopathologist. It also may require the input of the clinician who is at the bedside or in the office, especially in cases with extranodal involvement, in which the provision of clinical data to the pathologist is vital. The most salient examples of the need for collaboration are in ALK− ALCL, which may appear indistinguishable or difficult to distinguish under a microscope from primary cutaneous ALCL and other subtypes of CD30+ T- or B-cell lymphomas and classic Hodgkin lymphoma (as in our case), and large-cell transformation of mycosis fungoides. In both cases, the treatment may range from observation alone, to local radiation, to single noncytotoxic agents and single cytotoxic agents. The use of the term “large cell” does not always connote aggressive PTCL.
Conclusions
The aggressive PTCLs are a heterogenous group of uncommon lymphomas of mature T lymphocytes dominated by 3 subtypes: systemic ALCL, both ALK+ and ALK−; nodal PTCL with TFH phenotype; and PTCL-NOS. Although the accurate diagnosis of TCL and the subtyping of these lymphomas may be challenging, there is growing evidence that knowledge of the subtype of disease can aid in prognostication and in selection of optimal treatments, both in the front-line and in relapsed or refractory disease. The focus of this article has been 3 most common subtypes of aggressive PTCL as examples of how current knowledge may dictate choices in therapy and consultative referrals and inform rational targets and correlative studies in the development of future clinical trials. Finally, note that clinic-pathologic correlation, especially in the case of TCLs that may present with an extranodal component, is essential in the accurate diagnosis and subsequent treatment of these patients.
Correspondence
Lauren Pinter-Brown, Chao Family Comprehensive Cancer Center, 101 The City Dr S, Unit 23, Orange, CA 92868; e-mail: lpinterb@uci.edu.

