Abstract
The porphyrias are a family of metabolic disorders caused by defects in the activity of one of the enzymes in the heme biosynthetic pathway. Acute intermittent porphyria (AIP), caused by autosomal dominant mutations in the gene encoding hydroxymethylbilane synthase, can lead to hepatocyte overaccumulation and systemic distribution of the proximal porphyrin precursors, 5-aminolevulinic acid (ALA) and porphobilinogen (PBG). ALA and PBG are toxic to neurons and extrahepatic tissue and cause the neurovisceral clinical manifestations of AIP. Management of AIP includes awareness and avoidance of triggering factors, infusions of hemin for severe acute attacks, and, if indicated for chronic suppressive therapy, maintenance treatment with hemin or givosiran, a small interfering RNA molecule that antagonizes ALA synthase 1 transcripts. Erythropoietic protoporphyria (EPP) is most commonly caused by autosomal recessive mutations in the gene encoding ferrochelatase (FECH), the heme pathway terminal enzyme. FECH deficiency leads to erythrocyte overaccumulation and high plasma levels of lipophilic protoporphyrins that photoactivate in the skin, causing burning pain and erythema. Protoporphyrins excreted in the bile can cause gallstones, cholestasis, fibrosis, and ultimately liver failure. Management of EPP includes skin protection and afamelanotide, an α-melanocyte stimulating hormone analog that increases melanin pigment and reduces photoactivation. Liver transplantation may be necessary for severe EPP-induced liver complications. Because AIP and EPP arise from defects in the heme biosynthetic pathway, hematologists are often consulted to evaluate and manage suspected or proven porphyrias. A working knowledge of these disorders increases our confidence and effectiveness as consultants and medical providers.
Learning Objectives
Understand the pathobiological basis of acute intermittent porphyria (AIP) and erythropoietic protoporphyria (EPP), as related to genetic mutations affecting specific enzymes in the heme biosynthetic pathway and clinical manifestations caused by overaccumulation of harmful porphyrin precursors
Diagnose AIP or EPP and appropriately screen at-risk family members by using and accurately interpreting biochemical assays and genetic confirmatory tests
Manage AIP or EPP by using proper avoidance measures, effective pharmacologic interventions, and longitudinal supportive care, including monitoring and interventions for chronic end-organ complications
Case vignette 1
A 24-year-old woman is referred by her primary care provider for recommendations after recently being diagnosed with porphyria. Since age 16, she has had sudden-onset episodes of severe abdominal, chest, or pelvic pain associated with burning skin, migratory numbness, and mental “spacing out.” These episodes, which last anywhere from 3 hours to 7 days, may be spontaneous or triggered by prolonged sun exposure, spicy foods, chemical fumes, or menses. Numerous evaluations over the years have failed to identify a clear diagnosis, and elimination diets were only transiently beneficial. After the patient learned that her cousin had acute intermittent porphyria (AIP), she purchased a direct-to-consumer genetic test kit and subsequently submitted the raw genomic data to a third-party online service for more detailed interpretation. That report identified a single-nucleotide polymorphism in the hydroxymethylbilane synthase (HMBS) gene that was determined to be of unclear clinical consequence. The patient’s diagnosis of porphyria was based on this genetic interpretation and a 24-hour urine quantitative porphyrin assay, collected during an acute abdominal pain episode, that revealed isolated, 2-fold elevation in coproporphyrin. Urinary levels of all other porphyrin precursors, including porphobilinogen (PBG), were normal. She reports almost immediate resolution of her symptoms after receiving an infusion of hemin during her most recent acute abdominal pain episode.
Why should hematologists know about porphyrias?
The porphyrias are a family of metabolic disorders caused by defects in the activity of one of the enzymes in the heme biosynthetic pathway leading to overaccumulation and excretion of porphyrin precursors in hepatocytes or erythroid cells, leading to extrahepatic or extramedullary cellular, tissue, and end-organ injury. The heme biosynthetic pathway involves 8 enzymatic steps and 7 committed intermediate porphyrin precursors that result in a protoporphyrin ring that incorporates iron to form the heme molecule. Hemoglobin and myoglobin use heme moieties to bind oxygen, and a number of other enzymes contain heme prosthetic groups that react with molecular oxygen and participate in electron transfer reactions.
As clinical hematologists we understand that elevated free protoporphyrin and zinc protoporphyrin are useful biomarkers of iron deficiency and lead poisoning. We might remember that lead inhibits ferrochelatase (FECH), the enzyme that catalyzes the insertion of ferrous iron into protoporphyrin IX (PPIX), thereby allowing PPIX to incorporate zinc ions. We also learned that X-linked sideroblastic anemia is caused by mutations in the 5-aminolevulinate synthase 2 gene (ALAS2), which encodes the erythroid-specific isoform of the enzyme that initiates porphyrin production.
Thus, our foundational knowledge of heme production and function provides insight into the pathobiological features of the 2 most common congenital porphyrias: AIP and erythropoietic protoporphyria (EPP). Because hematologists are often consulted to evaluate and treat patients with suspected or proven porphyrias, understanding these disorders increases our confidence as consultants and our skills as medical providers.
A brief primer on relevant steps in the heme biosynthetic pathway
Erythroid precursors produce roughly three quarters of total body heme, and hepatocytes produce much of the rest.1 Heme biosynthesis begins with production of 5-aminolevulinic acid (ALA). This step is catalyzed by 5-aminolevulinate synthase 2 (ALAS2) in erythroid cells and by 5-aminolevulinate synthase 1 (ALAS1) in the liver and other tissues. ALA production is the major rate-limiting step, and, importantly, ALAS1 and ALAS2 are differentially regulated. Hepatic ALAS1 is activated by cytochrome P450 inducers such as smoking, alcohol consumption, calorie deprivation, numerous pharmacologic agents, and female hormones, particularly progesterone. Heme and glucose decrease the production and stability of ALAS1, and exogenous heme (in the form of pharmaceutical hemin) is a strong negative feedback inhibitor of porphyrin synthesis in the liver (Figure 1). By comparison, ALAS2 production is downmodulated during erythroid maturation via the GATA1 transcription factor. Post-transcriptional regulation of ALAS2 is primarily through an iron-responsive translational control element in the ALAS2 messenger RNA. At the terminal end of heme biosynthesis is FECH, an iron–sulfur cluster protein that is controlled by a number of transcription factors that are active during erythropoiesis. Iron deficiency and impaired iron–sulfur cluster biogenesis reduce FECH activity and heme formation.
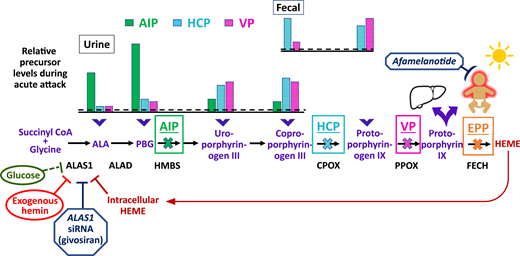
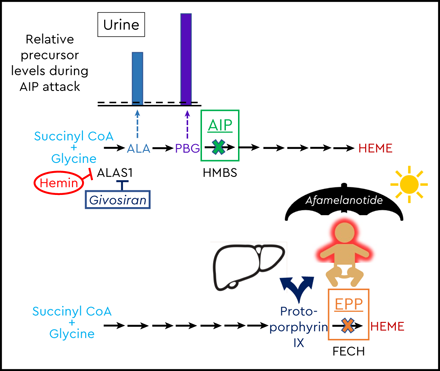
Schematic of the heme biosynthetic pathway with clinical correlates of three autosomal dominant AHPs: AIP, HCP, and VP; and autosomal recessive EPP. The 8 enzymatic steps (represented by arrows) and the key enzymes (protein abbreviations under arrows) highlight the pathobiological consequences of enzyme insufficiency. In patients with an AHP, induction of ALAS1 activity leads to buildup of the neurotoxic porphyrin precursors ALA and PBG, which distribute into tissues and cause neurovisceral signs and symptoms. The bar charts above the pathway depict the relative elevations of proximal and distal porphyrin precursors that are excreted into urine and feces with AIP, HCP, or VP during an acute symptomatic attack (dotted line represents relative 4-fold elevations above the upper ranges of normal). Relative fecal levels of coproporphyrin and protoporphyrin differentiate HCP from VP. Physiological inhibitors of ALAS1 include glucose and intracellular heme. Pharmacologic inhibitors include exogenous hemin and a hepatocyte-directed, small interfering RNA drug, givosiran, which targets the ALAS1 transcript. A fourth, extremely rare AHP is caused by ALAD deficiency. FECH mutations lead to erythrocyte buildup and leakage of PPIX. Lipophilic PPIX deposits in the skin, where it causes painful photosensitivity. Afamelanotide, which stimulates melanin production, can protect against photoactivation for patients with EPP. Biliary excretion of PPIX can lead to chronic cholestatic liver injury.
Schematic of the heme biosynthetic pathway with clinical correlates of three autosomal dominant AHPs: AIP, HCP, and VP; and autosomal recessive EPP. The 8 enzymatic steps (represented by arrows) and the key enzymes (protein abbreviations under arrows) highlight the pathobiological consequences of enzyme insufficiency. In patients with an AHP, induction of ALAS1 activity leads to buildup of the neurotoxic porphyrin precursors ALA and PBG, which distribute into tissues and cause neurovisceral signs and symptoms. The bar charts above the pathway depict the relative elevations of proximal and distal porphyrin precursors that are excreted into urine and feces with AIP, HCP, or VP during an acute symptomatic attack (dotted line represents relative 4-fold elevations above the upper ranges of normal). Relative fecal levels of coproporphyrin and protoporphyrin differentiate HCP from VP. Physiological inhibitors of ALAS1 include glucose and intracellular heme. Pharmacologic inhibitors include exogenous hemin and a hepatocyte-directed, small interfering RNA drug, givosiran, which targets the ALAS1 transcript. A fourth, extremely rare AHP is caused by ALAD deficiency. FECH mutations lead to erythrocyte buildup and leakage of PPIX. Lipophilic PPIX deposits in the skin, where it causes painful photosensitivity. Afamelanotide, which stimulates melanin production, can protect against photoactivation for patients with EPP. Biliary excretion of PPIX can lead to chronic cholestatic liver injury.
Classification and characterization of porphyrias
Acute hepatic porphyrias
Four disorders are associated with high systemic levels of ALA, leading to neurovisceral clinical manifestations and end-organ complications.1,2 Acute symptomatic attacks are triggered by physiological or exogenous “porphyrinogenic” factors that upregulate ALAS1 and flood the heme biosynthetic cascade with porphyrin precursors. AIP is an autosomal dominant (AD) disorder with a high estimated frequency in Whites, at roughly 1 in 2000, but with low penetrance (1%-10%) for symptomatic disease.1,2 AIP is caused by mutations in the hydroxymethylbilane synthase gene (HMBS), which encodes HMBS, the third enzyme of the heme biosynthetic pathway (also known as PBG deaminase). Defective HMBS can lead to accumulation of the proximal porphyrin precursors, ALA and PBG, that circulate systemically and saturate extrahepatic tissues to cause the various neurovisceral clinical manifestations of AIP (Figure 1).
Variegate porphyria (VP) is caused by AD mutations in protoporphyrinogen oxidase (PPOX). VP is less common than AIP except in South Africa, where 3 in 1000 Whites of Dutch ancestry are affected. This higher prevalence is related to a founder effect of immigrants from the 17th century. Of note, the incidence of VP in the Netherlands is similar to that in other European countries. Hereditary coproporphyria (HCP), caused by AD mutations in coproporphyrinogen oxidase (CPOX), affects roughly 1 to 2 in 1,000,000 Whites. Because the enzyme deficiencies with HCP and VP occur in the distal end of the heme biosynthetic pathway, the highest porphyrin precursor levels in the urine and feces are coproporphyrin and protoporphyrin, respectively (Figure 1). Different from AIP, the “downstream” lipophilic porphyrin intermediates that build up with HCP and VP deposit in the skin, where solar photoactivation can cause blistering and altered hair growth. ALA dehydratase (ALAD) porphyria is an extremely rare and severe acute hepatic porphyria caused by autosomal recessive (AR) mutations in the ALAD gene, which encodes the second enzyme in the heme biosynthetic pathway.
Photocutaneous porphyrias
Four disorders are associated with predominant skin signs and symptoms, with or without end-organ complications. The most common inherited disorder, affecting roughly 0.5 to 2.7 in 100,000 White children, is EPP.2 The majority of symptomatic EPP cases are caused by AR inheritance of a loss-of-function FECH mutation on 1 allele, with a common low expression FECH genetic variant on the other allele, resulting in FECH deficiency (<30%) (Table 1). Reduced FECH leads to excess red cell and plasma protoporphyrins that photoactivate in the skin, causing severe burning, erythema, and swelling. Protoporphyrins excreted in the bile can lead to cholestasis, gallstones, fibrosis, and liver failure.2 A less common inherited protoporphyria, X-linked protoporphyria (XLP), is caused by a gain-of-function mutation in the ALAS2 gene, resulting in high PPIX levels and a clinical syndrome identical to EPP.
Porphyria cutanea tarda is caused by acquired inhibition (in 80% of cases) or AD inherited deficiency of the uroporphyrinogen decarboxylase enzyme (the fifth step in the heme biosynthetic pathway). Suppression of uroporphyrinogen decarboxylase activity to <20% leads to skin deposition of uroporphyrin and heptacarboxyporphyrin, with resultant fragility, blistering, ulceration, and bullae. Congenital erythropoietic porphyria is caused by mutations in uroporphyrinogen-III synthase, which catalyzes the fourth step in the heme biosynthetic pathway.
Role of DNA testing for patients with suspected porphyria
Significant recent progress has been made in identifying pathogenic allele variants in heme synthesis genes that correlate with biochemical abnormalities and clinical phenotypes.3 Public databases, such as the Human Gene Mutation Database (www.hgmd.cf.ac.uk), provide catalogs of variant allele frequencies based on whole exome and genome sequencing. However, absence of important unpublished variants and inclusion of unvalidated variants limit their usefulness for porphyrias. For these reasons, the International Porphyria Molecular Diagnostic Collaborative was formed to classify and validate disease-specific genetic alterations for acute hepatic porphyrias (AHPs).4
Recently, direct-to-consumer (DTC) genetic testing has become a highly popular method to trace ancestry, assess disease risk, and inform wellness practices. People can also acquire their raw genotypic data from the DTC vendor for a web-based third-party company to query publicly available databases to identify additional genetic variants. The pitfalls with this practice include inadequate coding sequence coverage for many genes, technical errors, false positives, and misinterpretation.5 This issue is particularly relevant for people who believe they have an acute hepatic porphyria based on erroneous interpretation of DTC raw single-nucleotide polymorphism data,4 despite the lack of biochemical evidence and other clinical parameters, as illustrated in Case Vignette 1.
Genetic testing is not part of first-line evaluation for a patient with suspected porphyrias (Figure 2). For AHPs, genetic testing is important after biochemical characterization to confirm the diagnosis and identify the relevant pathogenic variant in the index case. Recommendations about DNA testing and the use of mutation analyses for confirmation and family screening have been published by the Porphyrias Consortium of the National Institutes of Health’s Rare Diseases Clinical Research Network6 and are available online (https://www.rarediseasesnetwork.org/cms/porphyrias). Not all sequence variants have been validated as pathogenic, and a small number of pathogenic mutations are not detected by gene sequencing; therefore, the biochemical profile of porphyrin precursors in urine, feces, and blood remains the standard for diagnosis.7 Once a pathogenic mutation is identified, targeted gene testing can be used to screen all first-degree relatives.
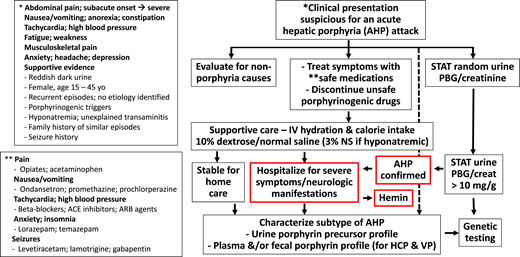
Diagnostic and intervention algorithm for patients presenting with signs and symptoms suspicious for an AHP attack. The neurovisceral manifestations of an attack with AIP are similar to those with HCP and VP. HCP and VP may also have a history of blistering skin rash on sun-exposed areas. A rapid spot urine quantitative assay for PBG can identify pathological accumulation and hyperexcretion of PBG and, by inference, ALA, the neurotoxic precursors that cause acute signs and symptoms. A urine PBG/creatinine ratio >10 mg/g is a sensitive and specific indicator of an AHP. Importantly, asymptomatic patients with AHP (particularly AIP) can have basal high urine PBG levels; therefore, it is always important to evaluate for alternative etiologies and porphyrinogenic triggers of acute signs and symptoms (eg, infection). Additional studies of urine, stool, and blood are needed to fully characterize the AHP subtype biochemically. Genetic testing is not appropriate for initial screening but is used to confirm the diagnosis based on biochemical testing results and can be very helpful for family screening. ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker.
Diagnostic and intervention algorithm for patients presenting with signs and symptoms suspicious for an AHP attack. The neurovisceral manifestations of an attack with AIP are similar to those with HCP and VP. HCP and VP may also have a history of blistering skin rash on sun-exposed areas. A rapid spot urine quantitative assay for PBG can identify pathological accumulation and hyperexcretion of PBG and, by inference, ALA, the neurotoxic precursors that cause acute signs and symptoms. A urine PBG/creatinine ratio >10 mg/g is a sensitive and specific indicator of an AHP. Importantly, asymptomatic patients with AHP (particularly AIP) can have basal high urine PBG levels; therefore, it is always important to evaluate for alternative etiologies and porphyrinogenic triggers of acute signs and symptoms (eg, infection). Additional studies of urine, stool, and blood are needed to fully characterize the AHP subtype biochemically. Genetic testing is not appropriate for initial screening but is used to confirm the diagnosis based on biochemical testing results and can be very helpful for family screening. ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker.
Case vignette 2
You are consulted on a 20-year-old woman admitted from the emergency department with a suspected acute porphyria attack. Since the birth of her first child 1 year ago she has had increasingly severe and more frequent episodes of gradual-onset abdominal pain with nausea, constipation, and occasional vomiting. The episodes occur at least monthly, usually before her menstrual cycle, and have also been associated with respiratory infections and heavy alcohol intake. Recent episodes have been more severe and prolonged, lasting 3 to 4 days, and associated with musculoskeletal pain, mental fogginess, distal paresthesia, headache, anxiety, and insomnia. Previous evaluations for gallstones, pancreatitis, appendicitis, and endometriosis have been negative. The symptom pattern has not improved with elimination diets, antacids, antiemetics, or hormonal manipulation with oral contraceptives. The patient takes trazodone for sleep and depression. She smokes tobacco and uses edible cannabis products. She reports that her 48-year-old mother was recently diagnosed with porphyria of unknown type. On presentation, she was afebrile, with blood pressure 160/100 mm Hg, pulse 100 bpm, and epigastric abdominal pain rated at 10 out of 10 severity. Abdominal examination revealed hypoactive bowel sounds with generalized tenderness on palpation but without rebound, guarding, organomegaly, or mass. Neurologic examination was notable for 4 out of 5 strength in the proximal arm muscles bilaterally. Laboratory data revealed a serum sodium of 128 mEq/L with otherwise normal electrolytes, renal function tests, and complete blood count. Liver function tests revealed mild elevations in alanine aminotransferase and aspartate aminotransferase, with normal amylase and lipase. A random urine semiquantitative assay for PBG was significantly elevated at 35 mg/L (normal 0–2.3 mg/L) with urine creatinine 1.7 g/L. Urine toxicology screen was positive for cannabinoids. The patient received an intravenous infusion of 10% dextrose overnight and hydromorphone for pain. Your examination confirms the abdominal and neurological findings noted in the ED without new deficits.
Clinicopathological features of AIP
Insights into the pathobiology of AIP have come from studies of patients who underwent orthotopic liver transplantation. These cases confirmed the liver as the source of PBG and ALA, the tissue-toxic porphyrin precursors. They also identified biomarkers of inflammation in liver explants along with elevated activity of heme oxygenase 1, which converts heme to biliverdin.8,9 A murine model of heterozygous AIP recapitulates these liver changes but also implicates hemin infusions in mediating inflammation and oxidative stress.9
The mechanisms responsible for acute and chronic neuronal and tissue injury with AIP are incompletely defined. ALA alone is sufficient to cause neurovisceral clinical manifestations, as exemplified by patients with rare ALAD porphyria, which is not associated with high PBG levels. A mouse model of severe homozygous dominant AIP suggests that ALA and PBG alter central nervous system (CNS) myelination and mediate neurotoxicity.10 At the cellular level, ALA affects the binding affinity of γ-amino butyric acid in neurons and compromises mitochondrial integrity and bioenergetics. ALA also causes vasoconstriction, presumably through an impaired oxidative stress response.11
Hyponatremia commonly accompanies AIP symptomatic attacks, and kidney disease develops in >50% of patients with symptomatic AIP.12 Patients with AIP also have increased risks of hepatocellular carcinoma and cholangiocarcinoma, which, hypothetically, may result from chronic inflammation, exogenous heme, and iron-mediated oxidative injury.13
Clinical presentation and chronic complications of AIP
The cardinal symptoms of AIP attacks are listed in Table 2. The “classic triad” of severe abdominal pain, peripheral neuropathy, and central or autonomic nervous system manifestations are variable and nonspecific and carry a broad differential diagnosis. Thus, diagnosis can be delayed for many years, and many patients are misdiagnosed or receive unhelpful treatments.7,14 Abdominal pain typically builds gradually over hours to days and is often diffuse and associated with nausea, vomiting, constipation, or diarrhea.14-16 Sensory neuropathy may be manifested as paresthesia, dysesthesia, hyporeflexia, and musculoskeletal pain in the extremities or trunk. Motor neuropathy often starts with proximal muscle weakness in the upper extremities and may progress to the distal and lower extremities. Respiratory muscle compromise can be life-threatening. Autonomic dysfunction is responsible for tachycardia, hypertension, diaphoresis, fever and chills, and bladder and gut dysmotility. CNS manifestations include insomnia, anxiety, depressed mood, dysphoria, confusion, delirium, seizure, and coma.
Historical and clinical features that support AIP as the cause of acute neurovisceral signs and symptoms are listed in Figure 2. Roughly 50% of women with symptomatic AIP report severe premenstrual symptoms, 42% experience exacerbations with oral contraceptive pills (OCPs), and 17% worsen during pregnancy.14 Patients with symptomatic VP and HCP present with similar acute neurovisceral manifestations as patients with AIP.15 A differentiating feature of VP and HCP (if present) is solar-activated skin pain and blisters with chronic pigmented areas and scars on sun-exposed areas.
Roughly two-thirds or more of patients with early adult-onset symptomatic AIP who experience recurrent attacks ≥2 times yearly develop by their fourth decade of life chronic disease complications with signs and symptoms that persist between acute episodes14-16 (Table 2). Quality-of-life parameters are also severely affected, with at least one-half of these patients reporting daily difficulties with pain, discomfort, anxiety, or depression. A significant portion are unable to perform usual activities or continue full-time work; and many experience economic hardship.15,16
Diagnosis of AIP
Patients with unexplained neurovisceral signs and symptoms suggestive of AIP should be questioned about hormonal, nutritional, or pharmacological triggering factors and family history supportive of the diagnosis. A publicly available database of porphyrinogenic drugs (https://porphyriafoundation.org/drugdatabase/) should be consulted to determine a possible causal association. Screening laboratory studies may reveal hyponatremia, mild transaminitis, red or brown urine (not related to hemoglobin or bilirubin), or leukocytosis without identifiable infectious, gastrointestinal, hepatobiliary, pancreatic, renal, or gynecologic cause. Because people with undiagnosed AIP may have multiple previous ED visits or take narcotics for unexplained severe pain episodes, they may be labeled as “drug seeking,” and urine toxicology screening studies are often obtained during initial evaluation. Such studies can be helpful to identify prescribed medications and nonprescribed substances of abuse.
The first-line screening test for a symptomatic patient with clinical manifestations suggestive of a neurovisceral porphyria attack is a quantitative assessment of PBG in a light-protected random urine sample. Urine creatinine should also be obtained to calculate a PBG/creatinine ratio. This will account for a possible spuriously low PBG result (ie, false negative) secondary to dilute urine from vigorous hydration. A substantially elevated random urine PBG/creatinine of >10 mg/g (>5 μmol/mmol) implicates an AHP (ie, either AIP, VP, or HCP) as the cause of the neurovisceral signs and symptoms.6,7 In the absence of a urine creatinine level, a spot urine PBG value >4 times the upper limit of normal (eg, >9.2 mg/L if the upper limit of normal is 2.3 mg/L) is significantly sensitive and specific to rule in a diagnosis of a neurovisceral porphyria. By comparison, a random urine PBG/creatinine level that is normal or only slightly elevated is strong evidence that neurovisceral porphyria is not the cause of the patient’s acute presenting signs and symptoms. Importantly, some patients with AIP persistently excrete high levels of PBG into the urine during asymptomatic periods between acute attacks. Therefore, nonporphyria etiologies (eg, gastroenteritis, peptic ulcer disease, biliary colic, pancreatitis, appendicitis, nephrolithiasis, urinary tract infection, pelvic inflammatory disease) should be considered, particularly if the presenting signs and symptoms are atypical.
To confirm the diagnosis and define the type of AHP for a patient with acute symptoms and a high urine PBG/creatinine ratio, full quantitative biochemical porphyrin precursor analyses should be performed on a properly collected (in appropriate buffer) urine sample at the time of an acute symptomatic episode, along with fecal and plasma porphyrin levels. The relative elevations of ALA, PBG, and more distal porphyrins provide clues to the etiology. Given the pitfalls of successfully collecting, handling, and processing a 24-hour urine sample for porphyria assessment, many centers have moved away from this previous approach. In AIP, ALA and PBG are markedly elevated, with lower relative increases in uroporphyrin, coproporphyrin, and protoporphyrin (Figure 1). Alternatively, marked elevations of the “downstream” porphyrins, with lower increases in ALA and PBG, suggest VP or HCP. The fecal and plasma porphyrin profiles can differentiate VP (with high protoporphyrin) from HCP (with high coproporphyrin).
Because full biochemical testing can be confounded by improperly collected 24-hour urine (ie, lacking the appropriate buffer, not light protected, or prolonged storage at room temperature) or collection after hemin infusion, repeat testing should be performed when in doubt. Ideally, this is done during an acute attack with typical signs and symptoms. False positive interpretation may also occur when the assay reveals <4-fold elevation of distal porphyrins, particularly an isolated mild increase in coproporphyrin alone.7 For suspected AIP, erythrocyte HMBS (PBG deaminase) enzyme activity assay is available; however, borderline normal to low levels may be seen in healthy people and patients with AIP. Thus, the HMBS activity does not add to biochemical characterization and genetic confirmation of AIP. Patients who seek consultation because of infrequent or remote neurovisceral symptoms with or without a family history could be screened with spot urine, fecal, and plasma porphyrin profiles. However, negative results mandate repeat testing during a symptomatic attack.
DNA testing for disease-causing mutations in HMBS, CPOX, and PPOX is not recommended for front-line screening.6,7 However, genetic screening may be considered in selected cases with a confirmed family history or personal history strongly suggestive of a neurovisceral porphyria.7 Genetic testing is otherwise indicated to identify the specific disease-causing mutations in biochemically confirmed symptomatic patients and for screening at-risk relatives of the index case.6,7
Management and monitoring of AIP
Management of acute signs and symptoms of a patient with known or highly suspected AIP involves recognizing the neurovisceral manifestations (and not overlooking other etiologies), treating precipitating factors (eg, occult infection), withdrawing porphyrinogenic medications, carbohydrate loading, symptom control, and supportive care including safe medications. Vomiting patients need intravenous hydration and may need correction of hyponatremia. Infusion of 10% dextrose, to deliver 300 to 500 g of glucose, may abort an early, mild attack; however, this is often not effective for more severe episodes and could worsen hyponatremia. For clinical complications severe enough to necessitate aggressive support in the clinic or ED, and with any new neurological deficit, intravenous infusion of hemin is indicated.1,2,6,7 Hemin should be reconstituted in 25% albumin and administered by slow infusion (eg, 60 minutes via peripheral intravenous line) to minimize coagulopathy and phlebitis.7 Patients who need frequent infusions of hemin benefit from placement of a semipermanent central venous catheter. To optimally suppress ALAS1 activity, 3 to 4 mg/kg/day for 4 days is needed. Although serial urine PBG/creatinine determinations during a course of hemin can reflect a biochemical response, clinical manifestations often do not correlate with absolute levels of urine PBG excretion. This is especially true for patients who chronically excrete high levels of urine PBG between episodes.
After nonporphyria etiologies of pain have been ruled out, escalating doses of opioids and anxiolytics can be carefully titrated while the patient is closely monitored for response and CNS side effects. Antiemetics and β-adrenergic blocking agents are useful for nausea and autonomic complications of tachycardia and hypertension. Levetiracetam and lamotrigine are safe antiseizure medications.
Longitudinal management of recurrent AIP episodes requires awareness and avoidance of factors that precipitate attacks, maintenance of a well-balanced diet with adequate carbohydrates (60%–70% total calories), a healthy lifestyle (eg, avoiding smoking and alcohol intake), and surveillance with management of the vascular and end-organ complications (Table 2). Some patients with frequent acute attacks benefit from scheduled maintenance infusions of hemin. These may be given up to twice weekly but require careful monitoring for benefit and tolerance.1,6,7 Complications include the need for central venous access, iron overload (which can be managed with phlebotomy), and a concern (based on animal models) that hemin may induce hepatic inflammation and potentially exacerbate clinical attacks.1,7,9
Women with symptomatic AIP and frequent cyclic neurovisceral attacks triggered by the luteal phase of the menstrual cycle may benefit from a low-dose combination oral contraceptive pill. However, this approach may worsen attacks in half of women and therefore must be carefully monitored. Standard-dose OCPs, progestin-only agents, implants, and intrauterine devices should be avoided. Alternatives include monthly preemptive infusions of hemin or ovarian suppression with a gonadotropin-releasing hormone agonist combined with a low-dose estrogen supplement to avoid menopausal symptoms and bone loss. Fertility is not impaired by AIP. Pregnancy exacerbates the frequency and severity of neurovisceral attacks in 15% to 20% of women. Hemin infusions can be safely given during pregnancy, when indicated, and are usually well tolerated. Ideally, a pregnant patient with AIP is co-managed by a high-risk obstetrician and, at the time of delivery, an anesthesiologist who is familiar with AIP and the contraindicated medications, potential complications, and interventions.
Chronic pain syndromes and neuropathy pose particular challenges. More severely affected patients benefit from co-management with a pain specialist. Multiple interventional approaches may be needed, including narcotics, gabapentin, antidepressants, and cognitive behavioral therapy. Psychiatric manifestations, including anxiety and depression, along with insomnia and fatigue significantly reduce quality of life and adversely affect family and social interactions.17 Safe antidepressants, anxiolytics, and other psychotropic agents may be used judiciously but must be closely monitored. Social service support is often needed because of the significant negative impact of recurrent AIP attacks on functional status and employability.16,17 For patients ≥50 years of age, liver ultrasounds and α-fetoprotein determinations are recommended every 6 months to monitor for hepatocellular carcinoma.6
Givosiran, a novel, liver-specific small interfering RNA molecule directed against ALAS1 messenger RNA, was approved in 2019 to treat patients with acute hepatic porphyrias and recurrent symptomatic attacks. Administered as a monthly subcutaneous injection, givosiran potently reduces ALAS1 transcripts and ALAS1 protein, thereby preventing overproduction of ALA and PBG during steady state and porphyrinogenic triggers. In the phase 3 pivotal trial, patients with AIP and a history of ≥2 symptomatic attacks in the preceding 6 months who received givosiran experienced a 74% reduction in the mean annualized rate of porphyria attacks and a 77% reduction in the mean annualized number of days of hemin, compared with patients who received placebo.18 In an extension phase of the trial, biochemical benefit was reportedly sustained out to 26 months. Givosiran can cause injection site reactions, nausea, and elevations in creatinine, amylase, lipase, and liver function tests, but was generally well tolerated. Importantly, givosiran interacts with CYP1A2 and CYP2D6 substrate drugs, and it is also a high-cost drug. Therefore, the optimal indications and value-based use of this agent for symptomatic patients with AIP remain to be defined.
Asymptomatic patients who carry a pathogenic HMBS mutation (identified through family screening) must be counseled to avoid porphyrinogenic triggers, particularly the broad list of medications and progestins that activate ALAS1 (https://porphyriafoundation.org/drugdatabase/). Formal recommendations for latent cases (ie, carriers with normal basal PBG excretion) and asymptomatic high excreters (ie, those with baseline urine ALA or PBG levels >4 times above normal) have been published.6 These include awareness of triggering factors, vigilance for signs and complications, healthy lifestyle practices, and a balanced diet without prolonged fasting or crash dieting. Because chronic high levels of ALA and PBG can induce end-organ damage in the kidneys and liver (with increased risk of hepatocellular carcinoma), asymptomatic high excreters should undergo thorough annual evaluations, including liver ultrasound and α-fetoprotein assessment when >50 years of age.6
Case vignette 2: follow-up
The high spot urine PBG/creatinine level was interpreted as diagnostic of AHP. The patient was aggressively treated with daily hemin infusions at 4 mg/kg/day for 4 days, and spot urine PBG/creatinine levels decreased significantly but did not normalize. Her symptoms improved, and she was stable for discharge after day 4 hemin infusion. Urine collected during the first hospital day revealed ALA and PBG levels 12- to 15 times above the upper limit of normal and uroporphyrin, coproporphyrin, and protoporphyrin levels 5- to 8 times above normal. Genetic testing revealed a mutation in HMBS (c.517C>T; p.R173W) that has been identified as a common pathogenic mutation in numerous unrelated probands. Family genetic studies confirmed inheritance from the patient’s mother. The patient subsequently needed monthly preemptive infusions of hemin to prevent cyclic AIP attacks, and she is under consideration for treatment with givosiran.
Case vignette 3
A 25-year-old woman seeks a second opinion on a diagnosis of porphyria. As a child, she lived in Hawaii and struggled with a “severe skin allergy” not responsive to antihistamines. By age 5, she was able to tell her parents that she had pain upon exposure to sunlight, and they asked her pediatrician test her for EPP. She was found to have “elevated blood porphyrins,” but no genetic testing was performed at the time. The family then decided to move to Oregon to help her shelter from the sun. During high school, she took oral β-carotene for 6 months, but she disliked the yellow skin discoloration and still could not withstand more than her usual 20 minutes of sun exposure without burning, swelling, and redness on her cheeks and hands that lasted for 2 to 3 days. She was recently offered a job at a prestigious company in southern California, but she is concerned that she cannot tolerate the sunnier environment. She has read on the Internet that there is a new treatment for EPP and is wondering whether it would be helpful.
Clinicopathological features of protoporphyria
EPP manifests with phototoxicity that happens when protoporphyrins in the superficial skin vasculature absorb light radiation and give off energy to form reactive oxygen species, resulting in lipid peroxidation, complement activation, and inflammation of the dermal capillaries.19 Almost 40% of patients with EPP report that a correct diagnosis required a mean of 12 years and evaluations by ≥5 physicians20 despite a similar percentage of patients presenting symptoms in the first year of life.21 Therefore, pediatricians should suspect EPP in children with unexplained allergic reactions, often labeled as “allergy to the sun.” A typical episode begins with burning and tingling sensation on skin exposed to the sun, developing into pain, swelling, and sometimes erythema. Pain can last ≤7 days and is not alleviated by medications, including opioids. Scarring, hyperpigmentation, hypopigmentation, and vesicles are uncommon, although repeated sun exposure leads to skin thickening and hyperkeratosis.19 Phototoxicity may be triggered by artificial fluorescent light and sunlight through windows, because UVA and visible light are not filtered. Patients eventually develop a sun avoidance behavior that affects their quality of life and social interactions.
Mild iron deficiency anemia occurs in 37% to 44% of patients. The etiology is unclear, but it may result from iron-mediated stimulation of ALAS2 production and excessive PPIX triggering inflammation that can block iron absorption.22 Excess protoporphyrins are excreted into the bile, accumulate in the liver, and cause cholestatic hepatitis, with severe disease in 2% to 5% of cases. Clinical hepatobiliary manifestations and gallstones usually develop after age 30. Table 3 compares the clinical presentations of EPP and XLP.
Diagnosis of protoporphyria
The investigation of suspected protoporphyria should include measurements of total, metal-free, and zinc erythrocyte protoporphyrins (ePPs).23 FECH deficiency with EPP leads to high ePP levels that are predominantly metal-free ePP (85% to 100%), with normal zinc ePP. By comparison, XLP, with preserved FECH and increased ALAS2 activity, leads to elevations in both metal-free (50% to 85%) and zinc ePP. Plasma protoporphyrins are also elevated, with a characteristic fluorescent peak at 632 to 634 nm. Notably, urinary protoporphyrins are normal. Of note, standard laboratories may not be able to provide reliable protoporphyrin levels; therefore, testing by a specialized laboratory is strongly recommended. Genetic testing establishes the diagnosis of EPP in the presence of two FECH gene mutations in trans, often including the hypomorphic variant c.315-48T>C (Table 1). Missense mutations are associated with lower ePP levels, longer tolerance to sun exposure, and a lower risk of hepatic complications. ALAS2 gain-of-function mutations confirm XLP. Next-generation sequencing has the potential to identify novel pathogenic variants among the 5% of patients with biochemical EPP who do not have mutations in either gene.24
Management of protoporphyria
Sun avoidance is the mainstay of management of phototoxicity. Sunscreen containing titanium or zinc oxide can block UVA and visible light but is cosmetically unpleasant. Tinted windows and use of protective clothing are helpful. The use of light filters (eg, yellow 61011 filters) during prolonged surgery or procedures can prevent skin and internal organ burns from fluorescent light exposure,25 particularly during liver transplant for cholestatic disease.
Oral β-carotene has been reported to benefit ≤40% of patients when used at therapeutic dosages that cause yellowish skin discoloration.21 However, multiple well-designed studies have failed to demonstrate efficacy. N-acetylcysteine, cysteine, vitamin C,26 and isoniazid27 have been tried without success.
Afamelanotide, an α-melanocyte–stimulating hormone analog delivered by a subcutaneous implant, was approved by the US Food and Drug Administration in 2019 for the treatment of EPP. It binds to the melanocortin 1 receptor and increases the production of eumelanin, providing photoprotection and antioxidant defense in melanocytes. In 2 randomized, placebo-controlled trials, afamelanotide prolonged pain-free time, decreased phototoxic reactions, and improved quality of life.28 The main side effects are nausea and application site reactions. Importantly, hyperpigmentation and increase in melanocytic nevi require full body skin examinations twice yearly. The maximum recommended regimen of 4 implants per year with an interval of 60 days represents a limitation for patients living in sunnier regions. Another stimulator of melanin production, MT-7117, is orally administered and now moving to phase 3 trials (NCT04402489).
Iron deficiency anemia with EPP is usually mild. Because iron can increase ALAS2 activity and worsen PPIX accumulation, iron supplementation should be prescribed only for patients with symptoms of iron deficiency. By comparison, preserved ferrochelatase activity in XLP facilitates iron incorporation into PPIX, and iron supplementation decreased photosensitivity in 7 of 8 female patients with XLP.21
Bone health is also a concern. Vitamin D deficiency, secondary to lack of sun exposure, affects 43% to 63% of patients with EPP, with elevated alkaline phosphatase and decreased bone mineral density.29 Physical activity should be encouraged, and smoking and excessive alcohol intake should be avoided.
Progressive hepatopathy with EPP has been managed with ursodeoxycholic acid, cholestyramine, and activated charcoal.1,2 Temporary use of red cell or plasma exchange with hemin infusions in cholestatic hepatitis is controversial.30 Liver transplantation has been performed in >50 published cases.31 However, a definitive cure can be achieved only with hematopoietic cell transplantation. It is usually considered after liver transplant for younger patients, for older patients with recurrent disease affecting the liver allograft, for progressive liver disease, or after reversal of liver failure without advanced liver fibrosis.32 A promising approach using gene therapy targeting ALAS2 in EPP erythroid precursors is under development.33
A summary of recommendations for monitoring of patients with EPP is shown in Table 4.
Case vignette 3: follow-up
Elevated total erythrocyte protoporphyrins (1155 μg/dL) with normal zinc protoporphyrins (55 μg/dL, reference value <60 μg/dL) yielded 95% of metal-free protoporphyrins, supporting the diagnosis of EPP. Genetic testing revealed coinheritance of the common variant FECH c.315-48T>C in trans with FECH c.1217G>A p. C406Y, a missense mutation. Liver function tests and a baseline abdominal ultrasound were normal. Complete blood counts showed iron deficiency anemia with hemoglobin 10.7 g/dL and ferritin 12 μg/L. Vitamin D level was low. Because the patient was asymptomatic, iron was not prescribed, but vitamin D supplements and a dual-energy X-ray absorptiometry scan were recommended. The patient was referred to a local specialist to start afamelanotide implants every 60 days. She will have full-body skin exams every 6 months and annual evaluations to monitor anemia, bone mineral density, and hepatobiliary status.
Correspondence
Michael Linenberger, University of Washington, 1100 Fairview Ave North, Mailstop D5-280, Seattle, WA 98109; e-mail: linen@u.washington.edu.