Abstract
Excellent outcomes in hematopoietic cell transplantation (HCT) from HLA-identical siblings, improvements in conditioning regimens, novel graft-versus-host disease prophylaxis, and the availability of alternative donors have all contributed to the increased applicability and acceptability of HCT for sickle cell disease (SCD). In young children with symptomatic SCD with an available HLA-identical related donor, HCT should be carefully considered. HCT from alternative donors is typically undertaken only in patients with severe symptoms, causing or likely to cause organ damage, and in the context of clinical trials. Patients undergoing HCT for SCD require careful counseling and preparation. They require careful monitoring of unique organ toxicities and complications during HCT. Patients must be prospectively followed for a prolonged time to determine the long-term outcomes and late effects of HCT for SCD. Thus, there is a need for a universal, longitudinal clinical registry to follow patients after HCT for SCD in conjunction with individuals who do not receive HCT to compare outcomes. Antibody-based conditioning and ex-vivo umbilical cord blood expansion are likely to improve the availability and acceptability of HCT. In addition, new disease-modifying drugs and the emerging option of the autologous transplantation of gene-modified hematopoietic progenitor cells are likely to expand the available therapeutic options and make decision-making by patients, physicians, and caregivers even more complicated. Future efforts must also focus on determining the impact of socioeconomic status on access to and outcomes of HCT and the long-term impact of HCT on patients, families, and society.
Learning Objectives
Discuss indications, conditioning, donor options, timing, outcomes, and decision-making in HCT for SCD
Understand the impact of recipient ages and the availability of HLA-identical donors on outcomes of HCT for SCD
Review emerging options in alternate-donor HCT for SCD.
CLINICAL CASE
A 12-year-old girl with HbSS-type sickle cell disease (SCD) has been having recurrent episodes of vaso-occlusive pain (VOE). She has been on hydroxyurea (HU) since the age of 9 months. Her clinical course remains severe despite an adequate trial of L-glutamine 3 years ago and, more recently, of voxelotor. She has an 8-year-old HLA-identical sibling. The patient's pediatric hematologist inquires whether she should talk to this family about considering hematopoietic cell transplantation (HCT).
Introduction
Comprehensive care and disease modification of SCD with HU can decrease morbidity and organ dysfunction and improve health-related quality of life (QoL) and survival.1 L-glutamine has been shown to reduce the rate of VOE and related hospitalizations.2 Voxelotor has been shown to increase the mean hemoglobin (Hb) level from baseline compared with placebo and may be particularly useful in individuals who have continued anemia and hemolysis.3 Crizanlizumab has been demonstrated to reduce the frequency of VOE.4 HCT, however, remains the only treatment with curative intent. When performed in young patients from HLA-identical related donors, HCT results in excellent overall survival (OS) and event-free survival (EFS).5-11 However, the lack of an available HLA-identical family donor remains a significant limitation in the applicability of matched sibling donor HCT. The optimization of supportive care, the development of novel conditioning regimens, and the availability of the options of HCT from alternative donors have enhanced the applicability of HCT for SCD. However, it is a sobering fact that, despite its increase in the last decade,11 HCT has been applied to only a tiny fraction of patients with SCD, including those with severe disease manifestations. This article aims to review the current status of HCT for SCD and address future directions in the field.
Considerations in decision-making about HCT for SCD
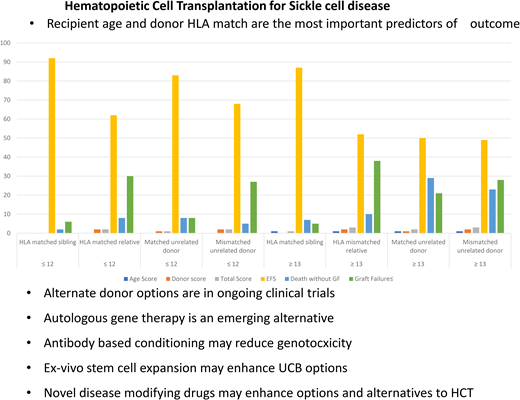
Recipient age and donor HLA match
The diagnosis of SCD is made at birth, and children are often well at a young age. However, the clinical course waxes and wanes and progresses unpredictably with age. Families can usually ascertain very early if their child with SCD has a potential HLA-identical sibling since siblings are often close in age. Thus, the crucial question is, at what age and when in the clinical course should HCT be considered? Gluckman et al9 reported in a study combining the European Society for Blood and Marrow Transplantation and Center for International Blood and Marrow Transplant Research registries that outcomes of HCT from HLA-identical related donors are excellent, but EFS decreases with increasing age at HCT (hazard ratio [HR], 1.09; P < .001). Cappelli et al8 reported 100% OS and 93% EFS in children under 5 years of age. Brazauskas et al12 performed an analysis of 1425 patients with SCD who underwent HCT between 2008 and 2017. They found that patients aged 12 or younger with an HLA-matched sibling donor had the best outcome, with a 3-year EFS of 92%.12 Age at HCT and type of donor were predictive of EFS. Patients ≤12 years undergoing HCT from an HLA-identical sibling are the best risk group. Patients ≤12 years receiving HCT from an unrelated donor and patients ≥13 years from an HLA-identical donor are at intermediate risk. All other patients are in the high-risk group (Table 1).
Indications for HCT
HCT for SCD has been performed in patients with severe SCD- related complications. Common reasons to proceed with HCT include a history of stroke, the need for chronic blood transfusions with the attendant risks of transfusional iron overload and features of disease severity, and predictors of premature mortality, such as recurrent VOE and recurrent acute chest syndrome.13-16 In patients enrolled in early clinical trials of HCT for SCD, the most common indications were stroke in 57% of patients and frequent VOE in 23% of patients.17 More recently, the most common indication for HCT has been recurrent VOE in over 70% of cases.9,18-20
The reasons for the shift in indications for HCT are unknown. It is possible that with the decline in the incidence of stroke,21 there are fewer patients with stroke present and considering HCT. It is also possible that a shift has occurred in the perception of patients, caregivers, and physicians regarding recurrent VOE being an appropriate indication for HCT.22-24 The number of hospitalizations or emergency room visits for SCD-associated pain has long been considered a surrogate measure of the total burden of pain. However, the frequency of health care utilization may be an inadequate measure of the daily burden of pain since many patients manage most of their pain at home.25 A hospital or emergency room visit represents a small fraction of the pain experience.26 Thus, some patients with a severe burden of pain may not meet eligibility criteria because they do not have frequent health care utilization. Acute intermittent vaso-occlusive pain is the hallmark of SCD, but more than half of the adults with SCD transition from acute intermittent pain to chronic persistent pain. Chronic pain, defined by duration as the presence of pain on most days of the previous 6 months, is a significant cause of morbidity and impaired QoL in SCD.27 However, chronic pain may not affect all individuals with the same degree of disability and interference with activities.
A definition of chronic pain based on duration alone does not consider the multiple dimensions of the condition or capture the extent of associated disability. The US National Pain Strategy has proposed high-impact chronic pain (HICP) as an extreme phenotype of chronic pain associated with severe disability.28 HICP, determined by screening patients for frequent daily pain and the presence of disability,29 is beginning to be used as an eligibility criterion for HCT for SCD even in the absence of frequent health care utilization.28,29 However, more research is required on how to integrate screening for HICP into clinical care and determine from electronic health records if an individual has HICP. Post-HCT patients with SCD show improvement in pain interference, opioid use, hospitalization, and QoL.18,30-33 A subgroup of patients with the pre-HCT features of significantly higher pain burdens, anxiety, and the use of long-acting opioids before HCT have persistent chronic pain beyond 1 year post HCT.33 Thus, the identification of HICP pre-HCT, careful preparation of patients, including behavioral health consultation, and long-term multimodal rehabilitation post-HCT are crucial to optimize outcomes in these patients.
Donor considerations
Type of donor
The majority of cases of HCT for SCD reported to international registries represent HLA-identical sibling donor HCT.11 Outcomes of HCT from HLA-identical donors are superior to those from alternate donors,11,34 but the availability of HLA-identical donor HCT is severely limited by the lack of suitable family donors.35 The use of alternate donors to expand the donor pool for patients with SCD HCT has been the subject of intensive investigation. HCT from unrelated donors results in stable donor-derived hematopoiesis but is associated with significant HCT-related morbidity and mortality.36 The addition of costimulatory blockade using abatacept for graft-versus-host disease (GVHD) prophylaxis has shown promise in mitigating the risk of severe GVHD in HCT for SCD and is the subject of an ongoing multicenter clinical trial (NCT 03924401).37 HCT from HLA-haploidentical familial donors using in-vivo or ex-vivo T-cell depletion has been reported to be safe and effective in early-phase clinical trials and is the subject of ongoing multicenter clinical trials (NCT 03263559, NCT04201210).20,38-41
Stem cell source
No study to date has compared the outcomes from HCT using different stem cell sources. A higher rate of nonengraftment led to the premature closure of the unrelated donor (URD) umbilical cord blood (UCB) arm of the BMT CTN 0601 study.42 A recent case series suggests that the addition of thiotepa to the reduced-intensity conditioning (RIC) regimen may enhance the engraftment of UCB.43 Peripheral blood as a stem cell source has been associated with a higher risk of chronic GVHD (CGVHD).11
Donor characteristics
The impact of donor age in HCT for SCD has not been reported. However, donor age has been reported as a risk factor for CGVHD following HCT for leukemia.44 Thus, donor age is likely significant since GVHD does not convey any benefit in HCT for SCD. Increasingly, patients with SCD are receiving HCT from familial haploidentical donors, such as parents or older siblings; hence, it is crucial to determine the contribution of donor age to outcomes. In addition, ABO major or minor incompatibility may add to the risk of graft stem cell loss,42 delayed red cell engraftment decreased OS [44], and pure red cell aplasia.45 Therefore, donor size is an important consideration to ensure an adequate cell dose without compromising donor safety. For pediatric donors for HCT for SCD, the usual practice is to accept a minimum donor size ≥ 10kg and age ≥ 1 year. Donor-recipient cytomegalovirus serostatus is matched (D−/R− or D+/R+) whenever possible to minimize cytomegalovirus disease risks. High-titer donor-directed HLA antibodies may predict an increased risk for graft rejection. Therefore, recipients should be screened for the presence of donor-directed HLA antibodies. If high-titer antibodies are found to be present, desensitization must be considered before proceeding to HCT.45,46
Conditioning regimen for HCT
Most often, matched related donor (MRD) HCT has been performed following a myeloablative conditioning regimen.9 While early case series reported the use of a myeloablative combination of busulfan and cyclophosphamide,17 more recent series describe the use of RIC.9,36,42,43,47,48 The most common RIC approach is substituting cyclophosphamide with fludarabine in combination with another agent,9 usually an alkylator such as busulfan or melphalan.48 Several reduced-toxicity conditioning regimens have been described, including the use of reduced doses of busulfan combined with fludarabine with or without cyclophosphamide or the substitution of busulfan with treosulfan or with the addition of thiotepa.18,49-52 Nonmyeloablative conditioning with 200 cGy of total body irradiation and fludarabine resulted in poor long-term engraftment.53,54 A combination of total body irradiation (300 cGy) with alemtuzumab resulted in high OS and EFS in adults and children.19,31,55,56 Pretransplant immunosuppression with 2 courses of fludarabine and dexamethasone to prevent graft failure has been piloted in haploidentical- and URD HCT for SCD.45,57
Prophylaxis for GVHD
The depletion of T cells in vivo using either antithymocyte globulin (ATG; 70.6%) or alemtuzumab (11.5%) has been used extensively in patients undergoing MRD HCT and may be important for the prevention of graft failure.9 Bernaudin et al reported that the use of ATG may decrease the rate of graft failure from 22.6% to 3%.58 ATG has not been frequently used in MRD UCB transplantation (UCBT).59 The depletion of T cells in vivo with alemtuzumab has been used in MRD HCT and URD UCBT.19,42,43,48 The depletion of T cells in vivo with posttransplant cyclophosphamide with ATG or alemtuzumab has also been used for haploidentical HCT for SCD.45,47,60-62 The depletion of T cells ex vivo with CD34+ selection,63 CD3/CD19 depletion,64 or α/β T-cell receptor and CD19 depletion has also been used in haploidentical HCT for SCD.65 Ex vivo α/β t-cell receptor and CD19 depletion may reduce the risk of GVHD but may be associated with delayed immune reconstitution and an increased risk of infection and graft failure.38,65 The most commonly used GVHD prophylaxis consists of calcineurin inhibitors (CNIs), which are often combined with methotrexate (MTX) or mycophenolate mofetil.9 Locatelli et al reported that the addition of MTX following MRD UCBT reduced EFS.59 Therefore, mycophenolate mofetil is substituted for MTX in MRD UCBT.42,43 The addition of selective inhibition of T-cell costimulation with abatacept can decrease GVHD and thus improve the safety profile and applicability of HCT to SCD.37
Cell dose considerations
For MRD UCB, a total nucleated cell (TNC) dose for UCB >3×107 TNC/kg recipient weight is preferable.66 If the UCB cell dose is low, a combination of UCB and bone marrow may be used.67 In URD UCB, TNC >5×107/kg increased engraftment and disease-free survival.68 A target cell dose of 4×108 to 5×108 TNC/kg for bone marrow and 4×107 to 5×107 TNC/kg for UCB (prethaw) is recommended.
Engraftment following HCT
Risk factors for graft failure include HLA mismatch, high titers of donor-directed HLA antibodies, the intensity of conditioning, or the presence of active infection at the time of engraftment.69
A combination of donor chimerism in the lymphoid lineage (CD3) and myeloid lineage (CD15 or CD33), total Hb level, and sickle cell hemoglobin (HbS) percentage are used to evaluate the robustness of engraftment and donor-derived hematopoiesis. Whole-blood donor chimerism of 11% to 74% may be associated with stable donor-derived erythropoiesis.70-72 Myeloid and lymphoid lineage-specific chimerism may provide additional information, but erythroid cell chimerism assays are still being evaluated in research studies.73,74 Stable myeloid chimerism of at least 20% to 25% is associated with stable donor-derived erythropoiesis.18,71,72 HbS >50% suggests the immanence of autologous recovery. If there is mixed or declining donor chimerism or an increasing HbS percentage, closer monitoring with more frequent assessments of donor chimerism may be necessary. Donor lymphocyte infusions are associated with a significant risk of GVHD, and their role in improving donor chimerism is unknown.
Most patients rejecting the allograft reconstitute autologous hematopoiesis,58,71 even in the case of alternative-donor HCT.18,36,43,60,71 Marrow aplasia or prolonged cytopenia are indications for an urgent salvage HCT.18 Consideration of a second HCT should be deferred for at least 6 months following a first HCT that has resulted in graft failure with autologous reconstitution.
Organ function considerations specific to HCT for SCD
Neurological
In the initial case series of patients who underwent MRD HCT, seizures and hemorrhagic stroke were observed in 30% of patients.75 The risk factors for neurological complications included (1) a history of prior stroke, (2) hypertension due to neurological and renal dysfunction exacerbated by CNIs and corticosteroid use, (3) hemorrhagic stroke in patients with preexisting cerebral vasculopathy with post-HCT thrombocytopenia,58 and (4) posterior reversible encephalopathy syndrome (PRES) (22%-34%).36,76 There is an increased risk of PRES in SCD patients that is exacerbated during HCT and may result in decreased OS and EFS.76,77 As compared to individuals matched for age, sex, and race, individuals with SCD have lower blood pressure (BP).78 In patients with SCD, BP above the 50th percentile for age may be associated with an increased risk of stroke.78,79 Careful monitoring of BP and aggressive management of hypertension are particularly important when patients are receiving both corticosteroids and CNIs since both drugs are likely to contribute to the development of hypertension. In BMT CTN 0601, a multicenter trial of URD blood and marrow transplantation (BMT) for SCD, there was a high incidence of PRES.36 The GVHD prophylaxis in this study included the use of prednisone through day +28. Essential precautions to prevent neurological complications such as seizures or PRES include (1) starting antiepileptic seizure prophylaxis before conditioning, especially if busulfan is used, and continuing seizure prophylaxis for the duration of CNI administration, (2) carefully monitoring and strictly controlling BP,17,75 with a target BP within 10% of the median for age and sex for SCD patients as described by Pegelow et al,78 (3) maintaining normal magnesium levels by magnesium supplementation,80,81 and (4) administering prophylactic platelet transfusions to maintain a platelet count >50 000/µL and red blood cell transfusion as needed to maintain an Hb level of 9 to 11g/dL. Post HCT, the recommendation is to consider obtaining brain magnetic resonance angiography/imaging (MRA/MRI) at 1 and 2 years after HCT and then every 2 years as clinically indicated in patients with a history of stroke, moyamoya pretransplant, PRES, or another neurotoxicity during HCT.82 An age-appropriate neurocognitive evaluation should be obtained at 1 year. It should be repeated every 2 years if there is a history of a neurotoxic complication during HCT.82
Cardiopulmonary
In adult SCD patients, the combination of echocardiographic tricuspid regurgitant jet velocity >3.0m/s and brain natriuretic peptide >160pg/mL is a strong predictor of premature mortality.83,84 Following successful HCT there may be an improvement of tricuspid regurgitant jet velocity.18 Post HCT, obtaining an echocardiogram annually for 5 years is recommended. In addition, an evaluation of cardiac iron by T2 MRI at 1 year after HCT should be considered in patients with moderate or severe iron overload at HCT.82
Unique infection risks
SCD patients undergo autoinfarction of the spleen variably with age and have impairment of splenic function and an increased risk of pneumococcal sepsis,85-87 which most often includes nonvaccine serotypes.88 Overall, the risk of infection in SCD patients is increased due to splenic infarction, defective opsonization of encapsulated organisms, impairment of T- and B-cell immune function, and the presence of infarcted devitalized bone.89 SCD patients may recover splenic function on HU following the institution of chronic transfusion or following successful HCT.90 However, patients who are older at the time of HCT and those with extensive CGVHD are at higher risk of poor post-HCT splenic recovery.91 While pneumococcal infections are rare following HCT for SCD,91 deaths due to overwhelming pneumococcal sepsis have been reported. Therefore, pneumococcal prophylaxis, careful monitoring of splenic function recovery post HCT, and timely reimmunization starting with conjugated pneumococcal vaccines 6 months post transplant are important for the prevention of serious pneumococcal infections.92
Management of transfusional iron overload
Patients may have transfusional iron overload prior to HCT and may have also received several transfusions of packed red blood cells with the HCT. Patients who have received a lifetime transfusion burden ≥10 transfusions and serum ferritin ≥1500ng/mL are evaluated pre-HCT for liver iron overload and liver fibrosis. MR of the abdomen is performed for quantification of iron. In addition, ultrasound or MR elastography may be performed to assess the degree of fibrosis. In patients with evidence of severe iron overload or liver fibrosis, a hepatology consultation and liver biopsy may be considered for determining the safety of proceeding with HCT. Patients with cirrhosis, bridging fibrosis, or active hepatitis are too high risk and may not tolerate an HCT conditioning regimen.93 In patients with baseline iron overload, the removal of excess body iron stores must be instituted with oral iron chelation, by monthly phlebotomy, or by a combination thereof after measurement of residual iron overload by serum ferritin and MRI.94-97 Iron chelation may be initiated post HCT when patients are off immunosuppression, have no evidence of GVHD or drug-induced liver damage, and are transfusion independent.
Renal
Serum blood urea nitrogen/creatinine and glomerular filtration rate or 24-hour creatinine clearance and SCD-associated proteinuria are critical considerations pre-HCT and follow-ups for recovery post HCT.82 A prolonged course of CNIs places patients at a high risk of renal dysfunction. Patients with SCD with prior complement-mediated vascular injury due to their underlying primary hemolytic disease and additional stressors during the HCT process may develop progressive endothelial injury and end-organ dysfunction.98 Case reports and small case series of transplant-associated thrombotic microangiopathy in patients with SCD and following HCT suggest the need to monitor transplant-associated thrombotic microangiopathy, control hypertension, consider alternatives to CNIs, and treat with eculizumab when appropriate.99-103
Gonadal damage and impaired fertility
HCT recipients for SCD are at high risk of gonadal damage and infertility.104 However, data are lacking on the patient and caregiver perspective on the importance of fertility in making decisions about HCT, the costs of and access to fertility preservation, and the emotional and psychological impact of gonadal damage on the long-term survivors of HCT.105 Sperm banking in males and oocyte cryopreservation in females are standard procedures that can mitigate the risk of infertility.106 Ovarian tissue banking is an acceptable fertility-preservation technique that is no longer considered experimental and is the only method to preserve fertility for prepubertal girls.106 Testicular tissue cryopreservation in prepubertal boys is under investigation.107,108 Fertility preservation procedures pre-HCT must also consider that HU may cause gonadal damage and have an adverse impact on male and female fertility in SCD patients.109,110
Medicaid coverage of fertility preservation is extremely limited both in the scope of benefits and the number of states that require such a benefit.111 For example, only 15 states require fertility preservation coverage in private insurance plans, and 5 states extend this benefit only to females. In addition, state statutes provide variable coverage based on marital status, diagnosis, length of fertility problems, and the monetary limit of the benefit.
Long-term and late effects of HCT
HCT can stabilize organ function and ameliorate manifestations of SCD; patients need systematic follow-up of monitoring SCD complications and the long-term and late effects of HCT according to established consensus guidelines.82,112 Further, most of the published literature on the late effects of HCT for SCD is based on HCT for malignancies. There is, therefore, a need for a universal, longitudinal clinical registry to follow outcomes after HCT for SCD.
Values and preferences of patients, families, and caregivers
The decision to undergo HCT for SCD involves making complex trade-offs between the promise of relief of disease manifestations, stabilization of organ function, halting of disease progression, and improvement of longevity and QoL, on the one hand, with substantial treatment burden and morbidity in the short term, and the risk of new long-term sequelae such as CGVHD, infertility, and subsequent malignancy, on the other hand.24 Many factors are involved in patient and caregiver decision-making for SCD.24,113,114 These include the unpredictable onset and progression of disease complications, the repeated need to seek health care, a poor QoL, a worsening of disease-related complications, the need to make major therapeutic decisions, and concerns about the long-term consequences of SCD. Families that are aware of and have access to HCT, have strong family support, and have an available HLA-identical sibling donor are more likely to consider HCT.113 Overall, families face decisional conflict at levels that result in decisional delays or uncertainties.114 The perception that the current disease burden has become unacceptable is a common consideration for HCT for SCD among patients and their physicians.22-24,113 The availability of an HLA-identical sibling donor may be viewed as providential by parents and is a significant factor when considering HCT. Some patients or caregivers will not consider HCT at any level of risk.115,116 Almost three-quarters of patients report being willing to take a modest risk of ≥ 5% mortality, whereas 57% are willing to take the ≥ 10% risk of GVHD. Following a successful HCT, SCD patients and caregivers do not report decisional regret, even with active CGVHD.113
The physician perspective on decision-making
Their past experiences and the outcomes of previous patients may influence how physicians respond in making recommendations to patients about seeking HCT consultation. For example, Bakshi et al, in a qualitative study of physician decision-making in disease-modifying therapy for SCD, found that the physician's perception of the severity of disease and the ability of a given patient to adhere to the medication and treatment regimen is also a significant consideration in their recommending HCT for SCD.23
Socioeconomic status and HCT for SCD
Racial and socioeconomic disparities may adversely affect access to and outcomes of HCT in minority populations.117,118 There is also a substantial regional disparity in donor search coverage, transplant procedures, hospitalizations, medications, transportation, and lodging provided by different states' Medicaid programs for HCT.119 Mupfudze et al have described the burden of navigating eligibility for Medicaid, the peer review process for various components and procedures for HCT, the difference between fee-for-service and comprehensively managed plans, and the burden that HCT poses to families seeking HCT for SCD.120 Patients enrolled in Medicaid had a lower EFS (HR, 2.36; 95% CI, 1.44-3.85; P = .0006) and a higher cumulative incidence of graft failure following HCT (HR, 2.57; 95% CI, 1.43-4.60; P = .0015) compared to privately insured patients with SCD.121 Registry data provide no clue to the factors contributing to worse outcomes among Medicaid-insured patients. African Americans, Hispanics, and individuals with Medicaid coverage have been demonstrated to have worse outcomes following treatment for hematological malignancies and following HCT.117,118,122-124 These findings provide the rationale for further study of the complex interaction of psychosocial functioning, health behaviors, racial and ethnic disparities, poor socioeconomic status, and health care disparities on outcomes of HCT in patients with SCD.117,118,122-124
Future directions
Despite progress in the conditioning regimens, donor options, GVHD prophylaxis, and outcomes of allogeneic HCT over the last 25 years, HCT has been applied to a tiny proportion of patients with SCD. Advances in several fields, including amelioration of the risk of morbidity, mortality, and long-term sequelae, are necessary to increase the applicability and acceptability of HCT for SCD. Antibody-based myeloablation approaches to transplant conditioning through the use of anti-c-kit antibodies, anti-c-kit antibody conjugated to the drug saporin, a ribosome-inactivating protein with potent cell-cycle-independent cytotoxic activity, and c-kit targeted chimeric antigen receptor T cells can reduce or eliminate the need for chemotherapy-based conditioning regimens.125-131 A nonchemotherapy conditioning regimen can further reduce acute morbidity related to mucositis and marrow aplasia, gonadal toxicity, and subsequent malignancy related to alkylators. The refinement of GVHD prophylaxis using posttransplant cyclophosphamide or costimulatory blockade with abatacept can reduce acute GVHD and CGVHD and their sequelae. UCB is limited by both the availability of HLA-matched donors and the limited cell dose of available UCB units. Ex-vivo expansion of the cell dose of UCB units is likely to make more and possibly better HLA-matched units available. Advances in stem cell expansion and stem cell engineering, including identifying critical cytokines, proteins, and small drug agonists, have led to clinical protocols for expanding hematopoietic stem cells and improved engraftment. URD UCBT with omidubicel, a nicotinamide-based, ex vivo-expanded UCB product, is associated with rapid engraftment but a high rate of GVHD in patients with SCD.132 Ongoing clinical trials can refine the ex-vivo expansion of hematopoietic stem cells, with future applications to SCD.133 Pharmacokinetics-directed precision dosing can improve the safety and efficacy of in-vivo T-cell depletion with alemtuzumab and myeloablation with melphalan.134,135
Impaired fertility is an unintended consequence of the conditioning regimen. Improving the acceptability of HCT for SCD will require advocacy with state governments to expand access to a fertility preservation benefit, remove the spousal requirement, expand the diagnostic criteria, and include the benefit in Medicaid plans.111 Further study of improving the awareness of, access to, and outcomes of HCT in underserved populations can help us better understand how to make HCT promptly available to a greater proportion of SCD patients who could benefit from the procedure.
The advances in HCT are occurring contemporaneously with explosive growth in clinical trials of novel disease-modifying therapies, on the one hand, and gene therapy strategies, on the other hand. How the interaction of these developments is likely to affect the future status of HCT in the management of SCD is unknown. Still, it is safe to say that we have embarked on a journey of increased and improved therapeutic options for patients with SCD.
CLINICAL CASE (continued)
This 12-year-old girl with HbSS with features of severe disease phenotype is at risk for the long-term complications of SCD. Her clinical course has remained severe despite a trial of HU, L-glutamine, and voxelotor. She has excellent risk factors for HCT from her HLA-identical sibling. I would recommend that the hematologist have a series of conversations with the family to educate them about HCT and then refer them to HCT consultation and explore research procedures for ovarian tissue preservation. If the family provides informed consent, we will proceed to HCT with a pretransplant transfusion to lower HbS. During HCT we will maintain target Hb and platelet counts by transfusions, carefully manage BP, and maintain normal serum magnesium levels to prevent PRES.
Conflict-of-interest disclosure
Lakshmanan Krishnamurti: no competing financial interests to declare.
Off-label drug use
Lakshmanan Krishnamurti: nothing to disclose.