Abstract
α-Thalassemia major (ATM) is a severe disease resulting from deletions in all 4 copies of the α-globin gene. Although it is usually fatal before birth, the advent of in utero transfusions has enabled survival of a growing number of children. Postnatal therapy consists of chronic transfusions or stem cell transplantation, similar to patients with β-thalassemia major. In this review, we discuss the experience with postnatal stem cell transplantation in patients with ATM, as well as the ongoing phase 1 clinical trial of in utero stem cell transplantation for this condition.
Learning Objectives
Review HCT for patients with ATM
Discuss optimal conditioning regimens for HCT in patients with ATM
CLINICAL CASE
A pregnant woman of Southeast Asian ancestry underwent routine prenatal ultrasound and was found to have a fetus with hydrops fetalis, manifesting as ascites and placentamegaly. She and her partner were both known to be carriers of 2 α-globin deletions, giving them a 25% chance of a pregnancy with deletion in all 4 α-globin genes (α-thalassemia major [ATM]). Given the concern for ATM in the fetus, the couple were counseled regarding their options for the pregnancy, including pregnancy termination or fetal therapy with intrauterine transfusions (IUTs). They opted for IUTs, which were performed starting at 23 weeks of gestation and repeated every 3 weeks until birth. The baby was born at term and stayed in the hospital for 2 weeks. Postnatal transfusions were given every 3 weeks. Neurological development was normal at the time of testing at 8 months.
Introduction
Epidemiology and genetics
α-Thalassemia is a recessively inherited hemoglobinopathy caused by mutations in the α-globin genes located on the short arm of chromosome 16. It is one of the most common monogenic disorders in the world, affecting approximately 5% of the population; prevalence is highest in China, Southeast Asia, the Middle East, India, and Africa.1 There is a rising incidence in the Western United States due to immigration patterns such that in California 1 in 10 000 newborns have a clinically significant α-thalassemia, and the rate of ATM is 0.2 per 100 000 state births.2 α-Thalassemia has varying degrees of severity depending on the number of deleted or mutated genes and remaining functional α-globin genes.
Carriers of this condition can have (a) 1 α-globin gene deleted/inactivated in each chromosome (α−/α−), known as the trans form of the α-thalassemia trait (common in people of African descent), or b) 2 missing/inactivated α-globin genes on the same chromosome (αα/−−), known as the cis form of the α-thalassemia trait (common in people of Asian descent).
There are 2 clinically significant forms: ATM, also known as α0-thalassemia or hemoglobin (Hb) Bart's hydrops fetalis, caused by deletion/inactivation of all 4 α-globin genes (−/−), and HbH disease, most frequently caused by deletion/inactivation of 3 α-globin genes (−/−α).3
Normal and abnormal developmental Hb and pathology of hydrops
Fetal oxygen exchange is accomplished by embryonic Hb (composed of 2 zeta and 2 gamma chains, or ξ2γ2) until 2 months of gestation (Figure 1); thereafter, fetal Hb (α2γ2) performs this function.1 A reduction of α-globin results in the aggregation of γ-globin tetramers in utero (Hb Bart's) and after birth, of β-globin tetramers (HbH). Both Hb Bart's and HbH have increased oxygen affinity, resulting in poor oxygen delivery; they cause the premature destruction of mature red blood cells (RBCs), leading to hemolysis, and damage maturing erythroid precursors, leading to ineffective erythropoiesis.5 While most patients with a 3 α-globin gene deletion do not have severe symptoms, some patients with nondeletional mutations, such as Hb Constant Spring, require chronic transfusions and may benefit from stem cell transplantation.6

Hb switching at the human α- and β-globin loci. Green, embryonic globins; blue, fetal globins; red, adult globins. Primitive erythropoiesis, derived from the yolk sac, is characterized by the expression of ζ-globin (from the α-globin locus) and ɛ-globin (from the β-globin locus). These are silenced at approximately 8 weeks' gestation. α-Globin then accounts for the entirety of the transcriptional output from the α-globin locus. At the β-globin locus, there is a switch to fetal globin (γ-globin) during fetal life and then a second switch to the adult β-globin. The predominant type of Hb corresponding to each developmental stage is shown below. Adapted with permission from King and Higgs 2018.4
Hb switching at the human α- and β-globin loci. Green, embryonic globins; blue, fetal globins; red, adult globins. Primitive erythropoiesis, derived from the yolk sac, is characterized by the expression of ζ-globin (from the α-globin locus) and ɛ-globin (from the β-globin locus). These are silenced at approximately 8 weeks' gestation. α-Globin then accounts for the entirety of the transcriptional output from the α-globin locus. At the β-globin locus, there is a switch to fetal globin (γ-globin) during fetal life and then a second switch to the adult β-globin. The predominant type of Hb corresponding to each developmental stage is shown below. Adapted with permission from King and Higgs 2018.4
ATM is the most severe form: patients have a prenatal onset of nonimmune hydrops fetalis as a result of heart failure induced by severe anemia. Embryonic Hb Portland (ξ2γ2) is the only functional oxygen-carrying Hb in these infants, which is short-lived, as the ξ-globin gene is then silenced, switching to the nonfunctional Hb Bart's. Extramedullary erythropoiesis with marked hepatosplenomegaly and an enlarged placenta are common. With few exceptions, ATM is fatal in utero or shortly after birth without IUTs to treat the anemia. In untreated patients, hydrops can lead to maternal complications such as anemia, polyhydramnios, preterm labor, and preeclampsia.3
Prenatal diagnosis and counseling
Couples who are carriers for α-globin deletions and are at risk of having a child with ATM should receive genetic counseling to review the natural history of the disorder and discuss prenatal genetic testing, reproductive options, and prenatal and postnatal treatment options and their risks. During pregnancy, a definitive fetal diagnosis can be obtained using chorionic villus sampling, amniocentesis, or fetal blood sampling.7 Given the necessity of IUTs to enable survival, early diagnosis (preferably prior to the onset of hydrops fetalis) is important. Although some infants have, reportedly, survived without IUTs, they are almost always born preterm and have neonatal complications due to the effects of untreated fetal hypoxia.5
IUTs and impact on clinical outcomes
Given the severity of ATM and the lack of therapeutic options universally available until recently, parents have most often elected to undergo termination of pregnancy. However, IUTs are now commonly performed for multiple fetal anemias and are increasingly used to treat fetuses with ATM. Although the initial IUT can treat the critically low anemia, subsequent IUTs are generally given every 3 weeks to maintain an appropriate Hb level.1 Most fetal centers start IUTs at 18 weeks of gestational age and follow protocols similar to those for treatment of isoimmunization,8 with low complication rates (1.2% per procedure, 3.3% per fetus).9 Since most fetuses are diagnosed after 18 weeks, the most common approach is an intravenous infusion through the umbilical vein. The protocol is to transfuse O negative blood with a high hematocrit so that the anemia may be corrected without volume overloading the fetus. Several reports indicate that this fetal therapy provides benefits during the perinatal and neonatal period: it reverses anemia, fetal growth restriction, and hydrops; decreases preterm deliveries; and contributes to infants having higher Apgar scores and a shorter duration of neonatal ventilation compared to untreated patients.5 Several case series have also demonstrated a long-term positive impact on growth and development.1,5,10,11 After birth, infants with ATM continue to require transfusions, using a protocol similar to patients with β-thalassemia major (BTM). Given the improving survival of patients with ATM, parents can be given the option of fetal therapy during a nondirective prenatal counseling session.
Allogeneic hematopoietic stem cell transplantation for ATM
Allogeneic hematopoietic stem cell transplantation (HCT) is currently the only available curative treatment option for patients with ATM. The underlying principle of allogeneic HCT in ATM is similar to that in other hemoglobinopathies and involves the replacement of recipient hematopoietic stem cells (HSCs) with donor HSCs, resulting in the production of donor-derived RBCs that do not harbor the α-thalassemia mutation.
In 1998 a 21-month-old girl with ATM received a bone marrow (BM) HCT from a matched sibling donor (MSD) after conditioning with busulfan (Bu), cyclophosphamide (Cy), and horse-derived antithymocyte globulin (hATG). Graft-versus-host disease (GVHD) prophylaxis included methotrexate (MTX) and cyclosporine A (CSA). Neutrophil engraftment was detected at day +17. No major HCT-related complications developed. Hb levels remained >10g/dL without blood transfusions despite the presence of residual host HSCs.12 Since this first case published in 1998, 14 other patients who received an HCT for ATM have been shared with the scientific community (Table 1): 5 patients (33%) did not receive IUTs, and all were born premature, with lower Apgar scores at 1 and 5 minutes, had evidence of hydrops (3 mild, 2 severe), and required longer intensive neonatal care. Ten patients (66%) received IUTs and consequently had less hydrops and were born at term (50%) or late preterm. Despite some patients experiencing a challenging initial neonatal course, with chronic transfusions, intensive care, and close follow-up they were able to be bridged to transplant. Ten patients (66%) received HCT at an age ≤24 months, with the youngest being 5 months of age. In total, 16 HCTs were performed; 2 patients had graft failure, 1 of which was salvaged with a second transplant, while the other continued on chronic transfusions. In 5 patients the choice of conditioning regimen was not reported (NR). Of the remaining, the majority received myeloablative conditioning, which in 8 patients (53%) was a Bu-based regimen along with other agents, including fludarabine (Flu) and Cy. With respect to types of donors and stem cell source, 4 patients received an MSD (3 BM, 1 NR), 1 received related mismatched umbilical cord blood cells (UCB), 4 received matched unrelated donor stem cells (1 BM, 3 peripheral blood stem cells [PBSCs]), 4 got mismatched unrelated donor HSCs (2 UCB, 2 NR), and 1 patient received αβ T-cell- depleted haploidentical PBSCs. Only 1 patient, who was 13 years old at the time of transplant—the oldest patient reported—died from transplant-related complications. Considering those patients that engrafted, all became transfusion independent. Chimerism data were reported in 13 patients, and approximately half of the patients (6/13) were found to have mixed chimerism and became transfusion independent, similar to those with full chimerism. From those patients with known iron overload prior to HCT (9 total; 1 mild, 4 moderate, 4 severe), only 1 patient was reported to develop severe and 1 patient to develop mild veno-occlusive disease of the liver (VOD). Of the patients who successfully engrafted and survived, 6/13 (53%) had no developmental delays reported (66% of whom had previously received IUTs), and 46% (6/13) had a mild delay. For 1 patient this was NR.1,12-20
In contrast to ATM, the results of allogeneic HCT for a closely related hemoglobinopathy, BTM, are widely reported and well studied in large patient populations. Patients transplanted after the year 2000 have had an outstanding 2-year overall survival (OS) and event-free survival (EFS) with the use of MSD (93% and 85%, respectively) and BM (91% and 83%, respectively).22 However, in recent years the use of Bu-based myeloablative regimens have significantly improved outcomes with alternative donors, and they are now comparable to those of MSD (5-year OS and EFS for MSD and HLA-matched unrelated donors of 89% vs 87% and 86% vs 82%, respectively).23 Evidence also supports that patients who receive a transplant before 2 years have the best outcomes, with a 2-year OS and EFS of 95% and 93%.24
Traditional myeloablative conditioning regimens such as Bu/Cy typically result in sustained donor myeloid engraftment; however, they are associated with significant potential toxicity that can result in organ dysfunction, such as VOD and treatment-related mortality (TRM). Therefore, the use of reduced-toxicity conditioning (RTC) regimens, with the goal of minimizing toxicities without jeopardizing engraftment, is an appealing option and a continued subject of investigation in the field. Studies done in patients with high-risk BTM showed that substitution of Flu for Cy or Bu/Flu-based conditioning regimens led to myeloablation but were associated with decreased toxicity compared to the conventional Bu/Cy regimens, hence the term RTC.22,25 Further strides toward improved safety profiles of RTC regimens have been made with the use of pharmacokinetic (PK) model-based dosing of conditioning agents, which allows for reduced toxicity while maintaining the regimen's efficacy. The drug's exposure, measured as a cumulative area under the curve (cAUC) over the treatment course, can be adjusted in real time using therapeutic drug monitoring. A drug exposure below a certain threshold carries an increased risk of graft failure, whereas an exposure above the desired range is associated with an increased risk of organ toxicity. In a study comparing PK-targeted Bu and Flu and conventional Bu and Cy, the former approach was shown to be less toxic while maintaining efficacy in pediatric HCT patients.26 In several nonmalignant diseases, including thalassemia, this exposure could be further reduced due to the acceptability of mixed but stable myeloid chimerism, which often leads to therapeutic benefit. More recently, our group has shown that a less toxic and even lower exposure to Bu with a cAUC of 70 mg·h/L was adequate for disease correction and achievement of durable full or stable mixed myeloid engraftment (chimerism of >20%) in pediatric nonmalignant conditions, with rare cases of severe Bu- related toxicity and GVHD.27 Further, studies aiming to understand the relationship between graft rejection and Flu dose in children undergoing HCT have been conducted to optimize the immunosuppressive element of conditioning based on which a Flu cAUC of 16 to 20 mg·h/L is deemed safe and effective to overcome the immune barrier while showing no benefit of increased exposure with respect to survival.28,29 In a recent study by Contreras et al,30 children receiving RTC for nonmalignant HCT, a total of 62 patients, were transplanted (23% had a diagnosis of hemoglobinopathies, 7, BTM, and 7, sickle cell disease [SCD]). Using targeted Bu and Flu in combination with alemtuzumab (2012-2018), the cumulative incidence of graft failure was 6.9% (none occurred in patients with an underlying diagnosis of hemoglobinopathies), the 3-year cumulative incidence of TRM was 3.4%, and the 3-year OS was 96.6%, with a low incidence of acute GVHD (aGVHD) grade 2 to 4 and chronic GVHD (7% and 5%, respectively). These results suggest that the use of targeted Bu and Flu offers a well-tolerated option for children with nonmalignant disorders to achieve sustained engraftment with a low incidence of transplant-related complications.30
The threshold chimerism needed to render transfusion independence following transplantation in patients with thalassemias is not clearly known. However, the limited data in BTM suggest that complete donor chimerism is not essential for sustained engraftment or for achieving transfusion independence, and this is thought to be due to ineffective erythropoiesis, which allows the donor RBCs to have a survival advantage. The persistence of residual host cells at >25% early following transplant (<60 days) was shown to be a risk for graft rejection, while a level <10% was deemed low risk for rejection.31 Patients with persistent mixed chimerism (PMC), defined as a stable mixed chimerism for >2 years, were able to remain transfusion independent even with >25% residual host cells, and their graft was functional and sufficient to lead to transfusion independence.31 This is further supported by evidence from split chimerism in patients with PMC, where Andreani et al.32 observed that the proportion of donor-derived cells was equally distributed in the different cell lines, both in the peripheral blood and BM, with the exception of the erythrocyte compartment. Despite the presence of few donor-engrafted nucleated cells, the erythrocytes were almost completely donor in origin (80%-100%). This enrichment of donor RBCs in the blood was not observed in erythroid precursors from the marrow, suggesting that the ineffective erythropoiesis presumably responsible for this phenomenon works at a later stage of erythroid development.32 Consequently, since ATM is also characterized by ineffective erythropoiesis due to the imbalance of α and β chains,5 it is likely that complete donor chimerism is not critical, and PMC would be sufficient to render transfusion independence following allogeneic HCT.31
Rationale for in utero transplantation in patients with ATM
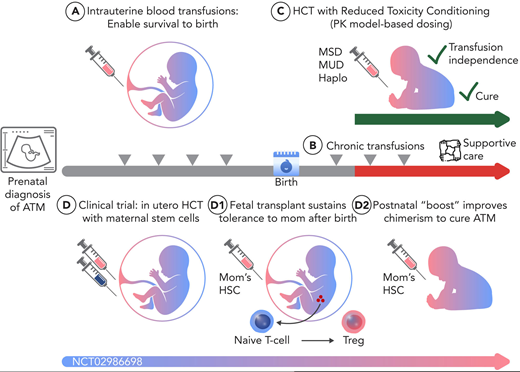
In utero HSC transplantation (IUHCT) is a promising therapy for diseases that can be treated by postnatal HCT. IUHCT takes advantage of the unique immune status in the fetus to allow engraftment of transplanted cells without conditioning.33 Our team is currently performing a phase 1 clinical trial of IUHCT (NCT02986698) using maternal stem cells as the donor: this strategy takes advantage of 2 aspects of maternal-fetal biology resulting from the natural trafficking of cells between the mother and the fetus: first, the presence of maternal cells in the fetus results in fetal tolerance to noninherited maternal antigens during pregnancy34 ; second, the maternal immune system can mediate rejection of third-party cells transplanted into the fetus in mouse models.35 Thus, transplantation of maternal cells provides the highest likelihood of success. We are currently enrolling patients with ATM for IUHCT between 18 and 26 weeks of gestation (Figure 2). It is important to note that although there are immune advantages to transplanting maternal stem cells into the fetus, the current lack of an appropriate conditioning regimen to create space in the hematopoietic niche results in low levels of engraftment in most animal models unless a very high dose of cells is infused. Therefore, the most likely scenario is that the prenatal transplantation is part of a 2-step protocol in which the in utero transplant achieves enough engraftment to sustain tolerance to maternal antigens after birth followed by a postnatal “boost” using maternal HSC with an RTC regimen with minimal or no immunoablation, improving engraftment to clinically meaningful levels. If successful, this protocol would overcome the lack of donor availability as well as offer an improved safety profile compared to traditional postnatal HCT. Indeed, promising outcomes have been seen in murine models of SCD and BTM in which IUHCT resulted in the development of donor-specific tolerance and PMC. A postnatal boost was given, and chimerism was further enhanced to high levels with near-complete Hb replacement (donor Hb >90%), ultimately correcting SCD and β-thalassemia phenotypes.36
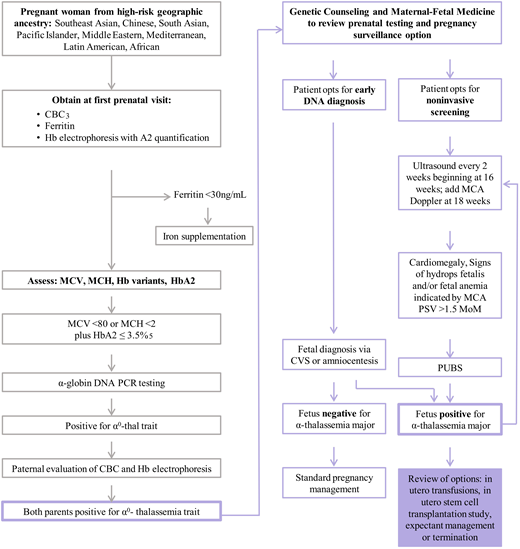
Prenatal screening for ATM. (1) This tool is not a replacement for referral to genetic counseling, which may happen at any time in this pathway. Genetic counseling provides guidance for genetic testing and management options for families with pregnancies at risk for severe forms of thalassemia. (2) The sensitivity and specificity are not definitive, and not all carriers will be detected by this screening. (3) The American College of Obstetricians and Gynecologists recommends that all pregnant women have a CBC with assessment of MCV. (4) Rare mutations, such as nondeletional α-thalassemia and others, may not be captured in this algorithm. In high-risk cases, or where Hb electrophoresis is abnormal, consultation with a genetic counselor and/or hematologist is recommended. (5) The presence of HbA2 > 3.5 does not exclude a coexisting α0-thalassemia trait. In individuals of Southeast Asian, Filipino, or Chinese descent who have microcytic hypochromic anemia, perform α-globin gene deletion and common variant studies irrespective of HbA2 level. CBC, complete blood count; CVS, chorionic villus sampling; HPLC, high-performance liquid chromatography; MCA, middle cerebral artery; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; MoM, multiples of the median; PCR, polymerase chain reaction; PSV, peak systolic velocity; PUBS, percutaneous umbilical blood sampling. Adapted with permission from the University of California, San Francisco hemoglobinopathy screening algorithm.
Prenatal screening for ATM. (1) This tool is not a replacement for referral to genetic counseling, which may happen at any time in this pathway. Genetic counseling provides guidance for genetic testing and management options for families with pregnancies at risk for severe forms of thalassemia. (2) The sensitivity and specificity are not definitive, and not all carriers will be detected by this screening. (3) The American College of Obstetricians and Gynecologists recommends that all pregnant women have a CBC with assessment of MCV. (4) Rare mutations, such as nondeletional α-thalassemia and others, may not be captured in this algorithm. In high-risk cases, or where Hb electrophoresis is abnormal, consultation with a genetic counselor and/or hematologist is recommended. (5) The presence of HbA2 > 3.5 does not exclude a coexisting α0-thalassemia trait. In individuals of Southeast Asian, Filipino, or Chinese descent who have microcytic hypochromic anemia, perform α-globin gene deletion and common variant studies irrespective of HbA2 level. CBC, complete blood count; CVS, chorionic villus sampling; HPLC, high-performance liquid chromatography; MCA, middle cerebral artery; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; MoM, multiples of the median; PCR, polymerase chain reaction; PSV, peak systolic velocity; PUBS, percutaneous umbilical blood sampling. Adapted with permission from the University of California, San Francisco hemoglobinopathy screening algorithm.
There have been multiple previous reports of IUHCT in multiple disease settings, including in fetuses with ATM with low engraftment.37,38 However, none of these previous reports has used the strategy of a high dose of maternal cells, infused intravenously, which is the protocol of our ongoing clinical trial for ATM. Of note, engraftment has been seen in the uniquely permissive setting of immunodeficiencies such as bare lymphocyte syndrome and severe combined immunodeficiency.39,40 We chose to start our clinical trial in patients with ATM since they require in IUTs for survival so there is no additional procedural risk from the transplantation protocol. If the protocol is safe, it could be applied to other hemoglobinopathies or other diseases that can be treated with HCT, such as Fanconi anemia or inborn errors of metabolism. Given the importance of cell source, dose, and other variables in engraftment, it is vital to perform IUHCT in the context of well-designed clinical trials.41
Conclusions and future directions
Although ATM was previously seen as a fatal disease, there are now multiple reports of patients who have survived to birth with excellent quality of life after IUTs. Since these patients still need chronic transfusions after birth, it is important to continue to develop strategies for a definitive cure using HCT. Postnatal HCT can be curative in ATM and can be safely performed using RTC, especially with PK model-based dosing to maintain efficacy while minimizing toxicity. It should be performed early in life to decrease the risk of TRM and other complications. In the future, nongenotoxic antibody-mediated conditioning regimens could further improve safety and efficacy.42 IUHCT is another promising approach that we are testing in a clinical trial. For IUHCT, it is important to use maternal stem cells to take advantage of the preexisting tolerance between the mother and fetus. This strategy will likely still require a postnatal “boost” transplantation, and defining safe and effective protocols to reduce transplant- related complications will be important. In the future, gene therapy or gene editing approaches may also be developed for these patients. Several strategies developed for β-thalassemia could be employed, including ex vivo lentiviral gene therapy to replace α-globin or gene editing to introduce α-globin into a genomic safe harbor. Given the improved survival of patients with ATM as a result of in IUTs, increasing numbers of patients with ATM will require chronic care: improved HCT or gene therapy options could be transformational in enabling definitive cures.
Conflict-of-interest disclosure
Paulina Horvei: no competing financial interests to declare.
Tippi MacKenzie: scientific advisory board member: Acrigen.
Sandhya Kharbanda: no competing financial interests to declare.
Off-label drug use
Paulina Horvei: nothing to disclose.
Tippi MacKenzie: nothing to disclose.
Sandhya Kharbanda: nothing to disclose.