Abstract
The application of genomic techniques, including cytogenetics and DNA sequencing, to decipher the molecular landscape of patients with myeloproliferative neoplasms (MPNs) has radically modified diagnostic approach and management through improved risk stratification. Three driver mutated genes (JAK2, MPL, CALR) are variably harbored by >80% of patients and associated with clinical characteristics, as well as major disease-related complications and different survival outcomes. Therefore, JAK2 V617F mutation is included in the revised International Prognosis Score of Thrombosis for Essential Thrombocythemia score for prediction of thrombosis in patients with essential thrombocythemia and prefibrotic primary myelofibrosis, while a CALR type 1 mutated genotype constitutes a favorable variable for survival in patients with myelofibrosis (MF). Novel, integrated clinical and cytogenetic/mutation scores (Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis [MIPSS70/v2], genetically inspired prognostic scoring system [GIPSS], Myelofibrosis Secondary to PV and ET- Prognostic Model [MYSEC-PM]) have been devised that guide selection of stem cell transplantation candidates with MF or help predict the risk associated with the transplant procedure (Myelofibrosis Transplant Scoring System), with greater performance compared with conventional scores based on hematologic and clinical variables only. On the other hand, several clinical needs remain unmet despite the great amount of molecular information available nowadays. These include the prediction of evolution to acute leukemia in a clinically actionable time frame, the identification of patients most likely to derive durable benefits from target agents, in primis JAK inhibitors, and, conversely, the significance of molecular responses that develop in patients receiving interferon or some novel agents. Here, we discuss briefly the significance and the role of genomic analysis for prognostication in patients with MPNs from a clinician's point of view, with the intent to provide how-to-use hints.
Learning Objectives
To appreciate the variety of abnormalities in the cytogenetic and mutation profiles of patients with myeloproliferative neoplasms (MPNs)
To be aware of the central role of molecular tests in the modern management of MPNs
To learn how to use at best molecular information for risk-stratifying patients with MPNs
To acknowledge the major clinical needs remaining unmet
Deep characterization of genomic abnormalities is essential for modern management of chronic myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), as well as post-PV and post-ET myelofibrosis (MF) (collectively, secondary myelofibrosis [sMF]). PMF includes an early/prefibrotic stage (pre-PMF) as well as an overt fibrotic stage, as 2 distinct diagnostic entities.1 Genomics informs diagnosis and risk assessment and supports therapy decision-making. Herein, we refer to genomic abnormalities in MPNs to include only changes in chromosomes/DNA sequence that deserve diagnostic and prognostic significance and can be identified through the application of methods available in specialized clinical laboratories. Therefore, this article is by no way an exhaustive review of genomics in MPNs, nor does it discuss mechanistic implications of genomic abnormalities.
(A few) Technical tips to know
Conventional methods to assess chromosomal abnormalities in MPNs are chromosomal banding (numerical and structural changes) and fluorescence in situ hybridization (that has the potential to discover also cytogenetically cryptic abnormalities), preferably using bone marrow (BM) or, if unavailable, peripheral blood (PB) cells. Driver mutations are routinely assessed in PB samples (preferably on gradient-purified granulocytes, which represent the variably involved myeloid clone and allow more reproducible measurement of mutation variant allele frequency [VAF] by eliminating contamination of lymphoid cells) by using different polymerase chain reaction–based techniques that, particularly for JAK2 V617F mutation, allow precise calculation of the ratio of mutated vs wild-type allele VAF; recommended assay sensitivity is ≤1%, in order to identify early disease presentation and, on the other side, to accurately monitor response to treatment (stem cell transplantation [SCT], interferon). Quantification of MPL and CALR VAF is not routinely performed owing to technical constraints and lack of standardized assays; however, distinction of type 1 and type 2 CALR mutation should be routinely reported since it is prognostically informative. Next-generation sequencing (NGS)–based methods are currently incorporated in the workup of selected patients with MPNs. Usually, they are panel based, interrogating 20 to 100+ most frequently mutated genes, with a conventional detection limit of 2% to 5%. Although NGS data can highlight structural changes and copy number variations, for clinical purposes, only single-nucleotide variants and insertions/deletions (indels) are reported. Ideally, attribution of a single-nucleotide variant to a somatic change requires paired analysis of a germline DNA source, which in clinical practice is unfeasible and expensive and is reserved for selected cases; therefore, somatic definition of a variant is based on comparative interrogation of large population databases to filter out known population polymorphisms. A second level of complexity in interpreting NGS data concerns the attribution of pathogenicity to the reported variant (ie, variant annotation), which requires bioinformatic expertise and should be performed in accordance with published guidelines.2
Chromosomal abnormalities in MPNs
Chromosomal alterations occur at varying frequencies and have variable prognostic significance depending on the underlying disease (Table 1). Cytogenetic abnormalities in PV and ET at diagnosis are rare (5%-15%) but increase with disease progression to sMF (up to 80%).3 Most common are deletion of the long arm of chromosome 20, trisomy 8, or trisomy 9. In PV, an intermediate- and high-risk karyotype category was identified (Table 1) influencing overall survival (OS) and leukemia-free survival.3,4 In ET, cytogenetic abnormalities are infrequent (7%) and associated with shorter survival (10-12 years vs 21 years without abnormalities).5 However, abnormal karyotype is not included in any integrated prognostic model for ET, while in association with advanced age, leukocytosis, and venous thrombosis, as well as advanced age and leukocytosis, abnormal karyotype is prognostically informative for OS and leukemic transformation, respectively, in PV.6
Conversely, specific chromosomal abnormalities are an integral component of prognostic scores for PMF, such as the DIPSS-plus, genetically inspired prognostic scoring system (GIPSS), and Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis (MIPSS70/v2) (discussed below). Patients with pre-PMF and overt PMF have a similar incidence and type of cytogenetic abnormalities.7 An isolated 13q−, 20q−, and 9+ abnormality deserves favorable prognostic value, while unfavorable abnormalities consist of a complex karyotype (>3 abnormalities) or a single or 2 abnormalities, including +8, −7/7q−, i(17)q, −5/5q−, 12p−, inv(3), or 11q23 rearrangements or a monosomal karyotype (MK). More recently, the prognostic value of cytogenetic abnormalities was refined in a series of 1002 patients, resulting in 3 risk categories8 : very high risk (VHR), favorable, and unfavorable karyotype (see Table 1 for details), with a median survival of 1.2 years (hazard ratio [HR], 3.8; 95% confidence interval [CI], 2.9-4.9), 2.9 years (HR, 1.7; 95% CI, 1.4-2.0), and 4.4 years (reference), respectively. The unfavorable and VHR karyotype conferred a 2.0- and 4.4-fold increased risk to progress to blast phase (BP), respectively.
An abnormal karyotype was found in 34% of 376 patients with sMF and had negative impact on median survival: 10.1 years (95% CI, 8.1-not reached) compared with 6.1 years (95% CI, 4.8-not reached) for normal karyotype.3 Shortened survival was further affected by the presence of a complex karyotype (2.7 years), complex karyotype without MK (3.4 years), and MK (2.1 years). However, when added to the Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) strata (see below), abnormal karyotype did not add to survival prediction.
Cytogenetic abnormalities constitute an important risk factor in the progression of MPNs to the accelerated phase (AP)/BP and are reported in up to 90% of patients.9 The chromosomal defects most significantly associated with AP/BP are the complex karyotype (including −5/del(5q), −7/del(7q), −17/del(17p)/i(17q), and −18), gains of chromosome 1q (MDM4 gene locus, encoding a negative regulator of TP53), or MK.10 Most patients with AP/BP show loss of 17p13, which leads to a deletion of TP53.11
Mutations associated with MPNs
MPN-associated driver mutations include JAK2 V617F mutation in >95% of patients with PV and 60% with ET and PMF; JAK2 exon 12 indels (the most frequent is N542_E543del) in 1% to 3% of JAK2 V617F–negative PV; mutations of the thrombopoietin receptor (MPL W515L/K/A) in 5% to 8% of patients with ET and PMF, including rare S505N; and exon 9 calreticulin mutations (CALR) in 20% to 25% of ET and PMF. There are 2 prototype CALR mutations: type 1 (52-bp deletion) and type 2 (5-bp insertion), with several other type 1–like and type 2–like variants. Driver mutations represent major diagnostic criteria in the latest World Health Organization (WHO) classification,1 as well as in the fifth edition of the WHO classification12 and the International Consensus Classification of Myeloid Neoplasms and Acute Leukemias.13 However, a driver mutation is missing in occasional patients with PV and 10% to 15% of those with ET and PMF (“triple negative”); this eventuality is acknowledged in current diagnostic systems that recommend NGS to search uncommon (“noncanonical”) somatic variants in JAK2 and MPL14 or to identify a clonal marker. NGS may also discover rare JAK2/MPL germline variants that underlie hereditary erythrocytosis or thrombocytosis mimicking MPNs.15,16
Similar to those with acute leukemia and myelodysplastic syndromes, patients with MPN may harbor mutations in a variety of so-called myeloid genes that include mainly epigenetic regulators, spliceosome components, and oncogenes (Table 2 lists the most frequent).17 These mutations may predate or follow the acquisition of the driver mutation and affect disease phenotype.18 Not all mutations have uniform prognostic significance. A set of so-called high mutation risk (HMR) genes,19,20 including ASXL1, EZH2, SRSF2, IDH1, IDH2, and U2AF1Q517, predict inferior survival in PMF, independent of each other and other risk factors, with >1 mutated gene harboring additional negative weight.21
CLINICAL CASE
A 42-year-old man presented to an outpatient clinic following the serendipitous discovery of thrombocytosis (800 × 109/L) in routine blood cell tests. Hemoglobin was in the lower range (13.2 g/dL with normal indexes), and leukocytes were 10.3 × 109/L, with no immature cells in the blood smear. All other routine tests were normal. He was asymptomatic, had no referred familial history for hematologic malignancies, and had no known generic cardiovascular risk factor; physical examination was unremarkable, and spleen was not palpable. An ultrasound scan confirmed normal spleen volume and also ruled out splanchnic vein thrombosis. The patient underwent BM biopsy, which revealed hypercellularity with granulocytic proliferation without atypia or blast increase; marked expansion of the megakaryocytic lineage with megakaryocytes of variable size and nuclei atypia, in tight clusters; and slightly reduced erythropoiesis. Reticulin fibrosis was grade 0/1. Cytogenetics showed a normal male karyotype. A JAK2 V617F mutation with a VAF of 34% was detected in PB granulocytes. On the basis of those findings, the patient received a WHO 2016 diagnosis of prefibrotic MF. The patient had a score of 2 according to the MPN-10 total symptom score (MPN-10).22
Molecular prognostication systems to address the risk of vascular events
Cardiovascular events are the main reason for morbidity and mortality in PV and ET; in pre-PMF, the rate of thrombosis is similar to ET, estimated at 1.99% patients/year. Patients with PV and ET are conventionally risk-stratified for thrombosis based on 2 clinical criteria: age ≥60 years and thrombosis history.23 However, the discovery that patients with ET who have a CALR mutation have a significantly reduced rate of vascular events led to the development of an integrated score, the International Prognosis Score of Thrombosis for Essential Thrombocythemia (IPSET), including the currently recommended revised version (Table 3).24 The positivity of a JAK2 V617F mutation, in the absence of the 2 aforementioned clinical criteria, qualifies a patient as low risk, with aspirin as suggested treatment. IPSET predicted thrombosis risk also in prefibrotic MF.25 The role of additional mutations for thrombosis in ET is largely unsettled.26 In PV, no compelling evidence for driver/additional mutations as being informative for thrombosis risk has been reported yet; the demonstration that a JAK2 V617F VAF ≥50% has an independent HR of 3.8 (95% CI, 1.7-8.6) for venous thrombosis27 might foster development of integrated scores.
Molecular prognostication systems to address the risk of dying
According to a series of 3023 patients with MPNs, median OS is around 20, 15, and 5 years, respectively, for ET, PV, and PMF.28 In ET, the conventional IPSET score uses age >60 years, leukocytes >11 × 109/L, and thrombosis history to differentiate low-, intermediate-, and high-risk patients with respective median survival not reached, 24.5 years, and 13.8 years.29 For PV, the International Working Group survival model delineated 3 risk groups with median survivals of 10.9, 18.9, and 27.8 years, based on older age, leukocytosis, and venous thrombosis.6 More recently, a mutation-enhanced international prognostic system (MIPSS) for ET and PV was devised.4 Spliceosome mutations adversely affected OS (SF3B1, SRSF2 in ET and SRSF2 in PV) and myelofibrosis-free survival (U2AF1, SF3B1 in ET); TP53 mutations predicted BP in ET. These adverse mutations occurred in 10% and 2% of patients with ET and PV, respectively. The integrated clinical-molecular model identified 3 risk categories with a median survival of 8.3 to 34.3 years in ET and 4.6 years to not reached in PV4 (Table 3). However, nowadays, these scores are not used in clinical practice for therapy decision-making.
Conversely, prognostic scores deserve a central role in the management of patients with MF specifically concerning the indication to SCT, the only potentially curative option; however, the not negligible risk associated with the transplant mandates careful selection of patients to make the procedure risk-effective.30 Conventional clinical-only scores, such as International Prognostic Scoring System (IPSS) and Dynamic-IPSS (DIPSS), or the cytogenetics-integrated DIPSS-plus continue to be largely used in practice and, notably, still are used for selection of patients in clinical trials. Furthermore, the IPSS and DIPSS scores are routinely applied also to patients with pre-PMF, although it was demonstrated that they poorly discriminate the intermediate 1 and 2 risk categories.7,31 Such shortcomings might be addressed by more recently developed, more informative, molecular-integrated scores.32
The abovementioned HMR genes configure an adverse variable in integrated risk scores for PMF such as the Mutation- Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis (MIPSS70)31 and MIPSS70v2.0, enriched with sex-adjusted hemoglobin and revised karyotype classification.33 MIPSS70, originally developed for patients of transplant age but informative age-independently as well, includes 9 variables, 3 genetic (HMR mutation, >1 HMR mutation, and absence of CALR type 1/like mutation), 5 hematologic-clinical factors, and bone marrow fibrosis grade ≥2 (Table 4). Based on unique variable-specific HR weighted scores, 3-tiered MIPSS70 low-, intermediate-, and high-risk categories, with corresponding median survival ranges of 27.7 years to “not reached,” 6.3 to 7.1 years, and 2.3 to 3.1 years, were identified and validated in 2 independent cohorts. Conversely, MIPSSv2 includes 5 risk categories: VHR (median survival, 1.8 years), high risk (4.1 years), intermediate risk (7.7 years), low risk (16.4 years), and very low risk (median not reached) (Table 4). Improved performance of MIPSS/v2 compared with conventional IPSS/DIPSS was demonstrated, with up to 40% of the patients being upgraded. Of note, the MIPSS score was developed for all patients with PMF and includes fibrosis grade (0-1 vs 2-3 category) as an embedded variable addressing the pre-PMF and overt-PMF category. A separate model based only on molecular factors, GIPSS, incorporated the 3-tiered karyotype categories and 4 mutations (ASXL1, SRSF2, and U2AF1Q157, plus absence of type 1/like CALR mutation) as independent risk factors for survival; risk categories were low (median survival, 26.4 years), intermediate 1 (8.0 years), intermediate 2 (4.2 years), and high (2 years [≥3 points]).34 For patients with sMF, the MYSEC-PM includes 5 clinical factors plus an unmutated CALR genotype, while the role of “myeloid” mutations is still debated.35 The simplified National Comprehensive Cancer Network guidelines stratification criteria include a lower and a higher risk category, based on variable scores of MIPSS and MIPSSv2, if genomics is available, or DIPSS-plus and MYSEC-PM if molecular testing is not available.36
Mutations are included also in the Myelofibrosis Transplant Scoring System (MTSS), an integrated score developed for predicting prognosis after SCT for PMF and sMF that includes age, Karnofsky performance status, thrombocytopenia, leukocytosis, human leukocyte antigen (HLA)-mismatched unrelated donor, ASXL1 mutation, and non-CALR/MPL driver mutation genotype. The 5-year survival was 83% (95% CI, 71%-95%), 64% (53%-75%), 37% (17%-57%), and 22% (4%-39%), respectively, in the low-, intermediate-, high-, and very high-risk categories37 (Table 4).
CLINICAL CASE (Continued)
The patient was categorized at diagnosis as IPSS low risk, with projected OS >10 years. No cytoreductive therapy was instituted, except low-dose aspirin since, based on the revised IPSET score, he belonged to the low-risk category. For the next 5 years, the disease course was uneventful. Then, his hemoglobin level began to downtrend (11.4 g/dL), mild leukocytosis (18 × 109/L) appeared steadily, platelets were 550 × 1012/L, 1% blasts and leukoerythroblastosis were detected in the blood smear, and lactate dehydrogenase was increased 1.8-fold above normal. Spleen was palpable at 4 cm from the left costal margin, and there was a recent onset of night sweats. The patient consulted another institution, where a new diagnostic procedure was performed. The bone marrow biopsy specimen revealed slightly reduced age-adjusted cellularity, with marked increase of atypical megakaryocytes in paratrabecular clusters; CD34+ cells were around 3%. Fibrosis was G2, with sparse areas of G3 and initial focal collagenization. PB blasts were 2%. Cytogenetics showed isolated trisomy 9. Targeted NGS panel identified mutations in ASXL1 (VAF, 24%) and SRSF2 (VAF, 33%). The VAF of JAK2 V617F had increased to 75%. A diagnosis of evolution of pre-PMF to overt PM was made. The disease was stratified as follows:
DIPPS-plus: intermediate 1 risk, due to constitutional symptoms and >1% PB blasts; the estimated survival is about 7 years.
MIPSSv2: very high risk, due to BM fibrosis ≥G2, constitutional symptoms, absence of CALR type 1 mutation, and HMR category with ≥2 HMR mutated genes; the estimated survival is <2 years with <5% likelihood of survival at 10 years.
The patient had become highly symptomatic and had an MPN-10 score of 44. An indication to hematopoietic SCT was posed after thorough discussion with the patient, based on MIPSSv2. He had a fully HLA-matched sibling donor. The MTSS score yielded an intermediate risk category, due to the absence of a CALR/MPL mutation and the presence of an ASXL1 mutation; the estimated 5-year survival was 77%. After SCT, the patient cleared the JAK2 V617F mutation at 4 months and HMR mutations (assayed at 1 year). He is alive, with no evidence of disease, after 3 years.
How we use molecular tests in patients with MPNs
Our approach to the use of molecular tests for prognostication purposes in MPNs is depicted in Figure 1. Driver mutations are stepwise interrogated in all patients since, beyond being required for diagnosis, they are prognostically informative in ET and pre-PMF for thrombosis (revised IPSET) and in MF for survival (favorable impact of CALR type 1/like). We use quantitative assays for measuring JAK2 V617F VAF at diagnosis and eventually document its increase at the time of evolution to post-PV and post-ET MF. Furthermore, serial measurement of JAK2 V617F might be informative in patients with PV receiving ropeginterferon to document changes in the clone of mutated cells.38,39 Owing to the limited applicability of survival prognostic models for decision-making, together with financial considerations, we do not currently support routine application of NGS to interrogate myeloid mutations in patients with PV and ET, while we perform NGS routinely in all transplant-age patients with PMF and sMF at diagnosis. Although most experience was obtained until now in overt PMF, we currently perform NGS also in younger patients with pre-PMF at diagnosis to calculate the MIPSS score, as the presented clinical case would have advocated. Results could be eventually used also for assessing the transplant-related risk (MTSS). There is still no indication if, and at what intervals, NGS should be repeated during the course of disease; in practice, nowadays NGS is usually performed coincident with some clinical and hematologic evidence of disease progression. In SCT-ineligible patients, integrated scores do not provide clinically actionable information, and we consider them not mandatory if cost considerations limit their use. Cytogenetics should be ideally obtained in all patients with MPNs at diagnosis and during the clinical course, if indicated, to exclude complex karyotype and/or TP53 involvement, although we acknowledge that results of the karyotype inform clinical decisions only in patients with PMF.
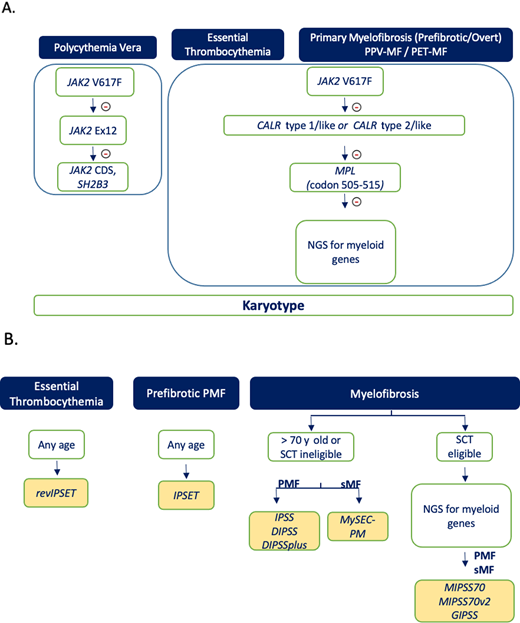
Flowchart on how we use genomic assays for diagnosis (A) and for prognostication in patients (B) with an established diagnosis of MPN. Selected information (cytogenetics, JAK2 V617F and CALR mutations) collected at the time of diagnosis may also inform risk scores at step B. NGS analysis for myeloid genes, if not available at the time of original investigation, is recommended in patients with PMF and sMF who are transplant eligible. PET, post-ET; PPV, post-PV.
Flowchart on how we use genomic assays for diagnosis (A) and for prognostication in patients (B) with an established diagnosis of MPN. Selected information (cytogenetics, JAK2 V617F and CALR mutations) collected at the time of diagnosis may also inform risk scores at step B. NGS analysis for myeloid genes, if not available at the time of original investigation, is recommended in patients with PMF and sMF who are transplant eligible. PET, post-ET; PPV, post-PV.
Conclusions
The past 10 years have witnessed incredible advancements in molecular-based prognostication of MPNs. Yet, we are left with a number of clinically relevant unmet needs, including the prediction of acute leukemia in a clinically actionable time frame, the prediction of response to JAKi (which might be adversely associated with RAS/CBL mutations),40 and the short- and long-term prognostic significance of molecular responses under treatment.38,41,42 However, “Nothing happens quite by chance. It's a question of accretion of information and experience” (Jonas Salk). So, it may be expected that discovery of new mutations, epigenetic markers, and altered RNA expression profile; application of a single-cell genotype; and exploitation of artificial intelligence to interpret genomic data might gradually result in novel, improved prognostic scores with a stronger impact on clinical decision-making for patients with MPNs.
Conflict-of-interest disclosure
Alessandro Maria Vannucchi has no relevant conflict of interest.
Paola Guglielmellihas has no relevant conflict of interest.
Off-label drug use
Alessandro Maria Vannucchi: nothing to disclose.
Paola Guglielmelli: nothing to disclose.