Abstract
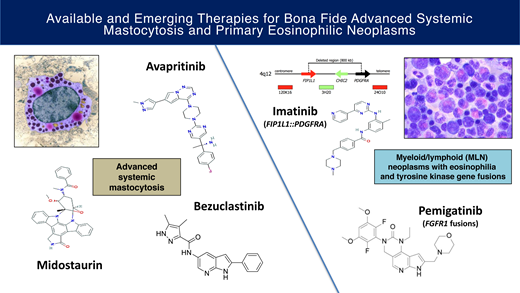
The historically poor prognosis of patients with advanced systemic mastocytosis (AdvSM) and primary eosinophilic neoplasms has shifted to increasingly favorable outcomes with the discovery of druggable targets. The multikinase/KIT inhibitor midostaurin and the highly selective KIT D816V inhibitor avapritinib can elicit marked improvements in measures of mast cell (MC) burden as well as reversion of MC-mediated organ damage (C-findings) and disease symptoms. With avapritinib, the achievement of molecular remission of KIT D816V and improved survival compared with historical therapy suggests a potential to affect disease natural history. BLU-263 and bezuclastinib are KIT D816V inhibitors currently being tested in trials of AdvSM. In the new World Health Organization and International Consensus Classifications, the category of “myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase (TK) gene fusions” is inclusive of rearrangements involving PDGFRA, PDGFRB, FGFR1, JAK2, FLT3, and ETV6::ABL1. While the successful outcomes with imatinib in FIP1L1::PDGFRA-positive cases and PDGFRB-rearranged neoplasms have become the “poster children” of these disorders, the responses of the other TK-driven neoplasms to small-molecule inhibitors are more variable. The selective FGFR inhibitor pemigatinib, approved in August 2022, is a promising therapy in aggressive FGFR1-driven diseases and highlights the role of such agents in bridging patients to allogeneic transplantation. This review summarizes the data for these approved and investigational agents and discusses open questions and future priorities regarding the management of these rare diseases.
Learning Objectives
Summarize the efficacy and safety data for targeted therapy of advanced systemic mastocytosis and primary eosinophilic neoplasms
Discuss the impact of these novel agents on current treatment algorithms and how they shape future management priorities
CLINICAL CASE
A 61-year-old man with systemic mastocytosis with chronic myelomonocytic leukemia (SM-CMML) presented with severe fatigue and a 30-pound weight loss over the prior 6 months. He did not respond to PEG-interferon-alfa-2a. For several months, his paracenteses requirement increased to twice weekly for a volume of 10 to 15 L. Examination was notable for temporal wasting and marked atrophy of muscles in the bilateral supraclavicular regions. Imaging findings of hepatosplenomegaly were not palpable on examination due to his marked ascites. Laboratory studies revealed a white blood cell count of 17.8 × 109/L (51% monocytes), hemoglobin of 11.9 g/dL, platelet count of 106 × 109/L, albumin of 2.3 g/dL (normal, 3.5-5.0 g/dL), and serum tryptase level of 416 ng/mL (normal, <11.5 ng/mL). The bone marrow biopsy specimen revealed 30% mast cells, increased monocytes, and 8% blasts/blast equivalents; karyotype revealed del(13q). Next-generation sequencing revealed KIT D816V and TET2 Q243X mutations (variant allele frequencies of 11% and 44%, respectively). The patient was referred for management.
Advanced systemic mastocytosis
Systemic mastocytosis (SM), defined by established World Health Organization (WHO) criteria, is divided into nonadvanced forms (indolent SM [ISM], bone marrow mastocytosis, smoldering SM) and advanced SM variants (aggressive SM [ASM], SM with an associated hematologic neoplasm [SM-AHN], and mast cell leukemia [MCL]).1-3 ASM is defined by ≥1 C-findings (neoplastic mast cell [MC]–related organ damage). Diagnostic criteria for SM-AHN include the presence of SM and an associated (myeloid) neoplasm (most commonly CMML),4 and MCL is histopathologically defined by ≥20% MCs on a bone marrow (BM) aspirate.1-4 Advanced SM (AdvSM) variants carry a poor prognosis (typically less than 4 years),5-9 and the worst outcomes are observed in patients with MCL (eg, <6 months to ~2 years).5,10-13 In both the International Consensus Classification (ICC) and WHO fifth edition,1,2 modifications to current minor diagnostic criteria include addition of CD30 as an immunohistochemical marker of SM, and an activating mutation in KIT besides D816V may also qualify. In the ICC,1 “AHN” is changed to “AMN” (associated myeloid neoplasm), and a core biopsy specimen may be used to diagnose MCL if the aspirate is a dry tap. While bone marrow mastocytosis is maintained as a subtype of ISM in the ICC,1 it is a distinct subtype from ISM in the WHO classification.2
Using high-sensitivity assays such as digital droplet polymerase chain reaction (PCR) and allele-specific PCR, KIT D816V can be detected in ~95% of patients with AdvSM.14,15 Compared with nonadvanced SM, AdvSM variants are more frequently characterized by multilineage involvement by KIT D816V and a complex, multimutated genetic landscape, best exemplified by SM-AHN.16-18 SRSF2, ASXL1, and RUNX1 (S/A/R panel) are high-risk mutations,19,20 but other myeloid mutations are commonly identified (eg, TET2, DNMT3A, JAK2, NRAS, CBL, EZH2),15 some of which have been incorporated into prognostic scoring systems based on clinical, laboratory, and molecular features.6-9
Historically, therapy of AdvSM has consisted of off-label use of PEG-interferon (IFN) α ± corticosteroids or cladribine. Studies evaluating these agents have often consisted of an admixture of patients with nonadvanced and AdvSM with overall responses in the range of 30% to 50% using heterogenous response criteria and variable reporting of biomarkers of response, including changes in BM MC burden and serum tryptase level.21-24 Imatinib was approved by the US Food and Drug Administration (FDA) in 2005 for patients with ASM without KIT D816V or unknown mutation status, an exceedingly rare patient population. While exon 17 KIT D816V is an imatinib-resistant mutation, the drug can be effective in patients with SM with exon 8 to 11 mutations/juxtamembrane variants such as F522C, which is associated with a well-differentiated SM phenotype.25 Acute myeloid leukemia (AML)–type induction chemotherapy has been used in patients with kinetically aggressive or refractory/relapsed disease to the aforementioned therapies. For patients with SM-AHN, in whom AHN-directed therapy is required, hydroxyurea has been used to control leukocytosis and splenomegaly. Hypomethylating agents are commonly employed when the AHN is a higher-risk myelodysplastic syndrome (MDS) or MDS/myeloproliferative neoplasm (MPN). A retrospective analysis of allogeneic hematopoietic stem cell transplantation (HSCT) revealed a 3-year overall survival of 57% among 57 patients, with a diagnosis of MCL and reduced intensity (vs myeloablative) conditioning being identified as adverse prognostic factors.26 These data were derived before the use of KIT inhibitors; therefore, the role of transplant in the modern KIT inhibitor era is not well defined.
The rationale for KIT inhibition in AdvSM is based on the high frequency of the KIT D816V mutation and its presence in cells from both the MC and AHN compartments.14,15,27,28 The experience with midostaurin and avapritinib has validated this clinical strategy but has also revealed challenges of KIT inhibitor monotherapy. In particular, patients with SM-AHN and/or cases with a complex molecular landscape are vulnerable to AHN progression or transformation to secondary AML.4,5,22-24
Midostaurin
Midostaurin (Novartis) is a multikinase/KIT inhibitor that targets not only D816V-mutated KIT but also wild-type (WT) KIT, PDGFRα/β, VEGFR2, and FLT3.29 Based on an encouraging partial response to midostaurin in a patient with SM-MDS/MPN-unclassifiable30 and an overall response rate (ORR) of 69% in an investigator-initiated trial,31 the drug was evaluated in a global registrational, phase 2, single-am, open-label study of 89 evaluable patients with AdvSM with ≥1 C-findings. The ORR was 60% (45% major responses) by (modified) Valent and Cheson criteria.32 The European Medicines Agency (EMA) and FDA performed post hoc analyses of the trial using International Working Group–Myeloproliferative Neoplasms Research and Treatment-European Competence Network on Mastocytosis (IWG) response criteria. The EMA and FDA identified IWG ORRs of 28% and 17%, respectively, the difference reflecting the FDA's decision not to include the category of “clinical improvement” in their calculation of response rate.33,34 Midostaurin elicited a median best percent reduction of serum tryptase level and BM MC burden by −58% and −59%, respectively; reduced spenomegaly; and elicited a significant reduction in symptoms (using the Memorial Symptom Assessment Scale) except for nausea and vomiting, which are common midostaurin-related adverse events.32,35 The median progression-free survival was 14.1 months, and the overall survival (OS) was 28.7 months.32 These data led to midostaurin's approval for advanced SM in 2017 by the FDA and EMA. Additional real-world cohorts of AdvSM patients have corroborated midostaurin's activity, including an analysis that showed that a ≥25% reduction in KIT D816V RNA expressed allele burden was the strongest variable associated with prolonged survival.36-39 A retrospective registry analysis also confirmed the superior survival of midostaurin compared with cladribine.40
Avapritinib
Avapritinib (BLU-265; Blueprint Medicines) was designed as a highly selective, type 1 inhibitor of D816V-mutated KIT.41 It exhibits a 10-fold lower 50% inhibitory concentration (IC50) compared with midostaurin in an assay of KIT D816V kinase activity (0.27 vs 2.9 nM). Avapritinib's restricted target profile also includes platelet-derived growth factor receptor alpha (PDGFRα) with negligible activity against WT KIT; the drug received FDA approval in 2020 for adults with an unresectable or metastatic gastrointestinal stromal tumor harboring a PDGFRA exon 18 mutation, including the D842V mutation.42
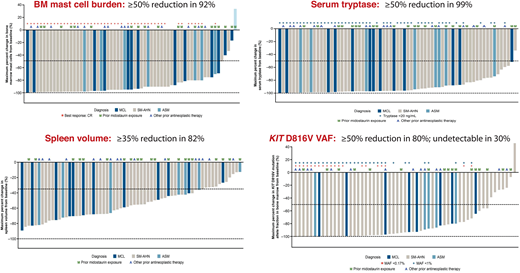
Avapritinib's FDA approval in 202143 as first-line therapy for adults with AdvSM (recommended platelet count ≥50 × 109/L) was based on the phase 1 EXPLORER study44 and an interim analysis of the phase 2 PATHFINDER study.45 The centrally adjudicated EXPLORER study consisted of a dose escalation phase evaluating doses of 30 to 400 mg daily in patients with AdvSM with ≥1 eligible organ damage finding by IWG criteria. The subsequent dose expansion phase evaluated 2 dosing cohorts of 200 mg and 300 mg daily. Ultimately, 200 mg daily was chosen as the recommended phase 2 dose based on a composite analysis of safety/tolerability, pharmacokinetics, efficacy, and biomarkers of response, including dynamic changes in BM MC burden and serum tryptase levels. Table 1 shows the overall rates of response by modified IWG criteria, AdvSM variant, prior therapy and midostaurin exposure, and S/A/R mutation status in the EXPLORER study (n = 69 evaluable patients)44 and from an interim analysis of PATHFINDER (n = 32 evaluable patients).45 Figure 1 highlights the marked decreases in measures of MC disease from the EXPLORER study, including BM MC burden, serum tryptase level, and KIT D816V variant allele fraction from BM using digital droplet PCR with a limit of detection of 0.17%. Importantly, a complete molecular remission (CMR) was achieved in 30% of patients, which represents a new response benchmark for AdvSM. Reversion of MC skin lesions and significant improvement in symptoms using the AdvSM Symptom Assessment Form were also observed.44-46
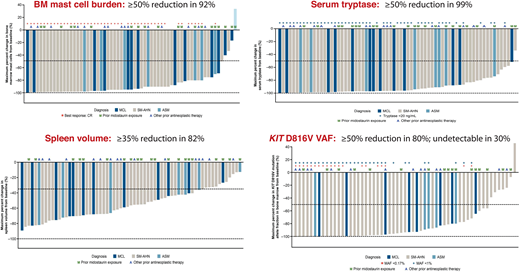
Phase 1 EXPLORER Study: Avapritinib effects on measures of mast cell burden. Waterfall plots are shown for bone marrow mast cell burden, serum tryptase level, spleen volume, and KIT D816V variant allele fraction. A, other antineoplastic therapy; M, prior midostaurin exposure; MAF, mutant allele fraction; VAF, variant allele fraction.
Phase 1 EXPLORER Study: Avapritinib effects on measures of mast cell burden. Waterfall plots are shown for bone marrow mast cell burden, serum tryptase level, spleen volume, and KIT D816V variant allele fraction. A, other antineoplastic therapy; M, prior midostaurin exposure; MAF, mutant allele fraction; VAF, variant allele fraction.
An analysis of pooled outcomes in 53 patients treated with an avapritinib starting dose of ≤200 mg daily revealed an ORR of 72% (complete response [CR]/CR with partial hematologic recovery [28%] + partial response [PR] [28%] + clinical improvement [15%]).47 The ORR among the 31 patients who had received prior therapy was 71%, including a CR/CR with partial hematologic recovery rate of 19%.48 Overall survival at 12 and 24 months was 80% and 65%, respectively, and the median OS survival was not reached in this previously treated population with a median follow-up of 17.7 months.48 In 2022, the EMA granted approval for avapritinib in patients with AdvSM exposed to at least 1 prior systemic therapy.49
Long-term outcomes
The estimated progression-free survival rates were 84% at 12 months and 63% at 24 months in the EXPLORER response-evaluable population (n = 53).44 During a median follow-up of 23 months, 14 (20%) exhibited disease progression, including 6 patients (9%) with transformation to secondary AML. There was no consistent pattern of baseline or on-treatment myeloid mutations, changes in the variant allele frequency of specific genes, or resistance mutations in KIT in patients with or without clinical progression.50
In the EXPLORER overall AdvSM safety population, the median OS was not reached with a median follow-up duration of 23 months.44 Patients with S/A/R mutations or a baseline mutation-adjusted risk score ≥2 exhibited shorter survival.44 Among all patients with AdvSM, the estimated 24-month OS rate was 76% and 100%, 67%, and 92% for ASM, SM-AHN, and MCL subtypes, respectively.44 The comparative survival rates from the global midostaurin trial were 53% for all patients with AdvSM and 86%, 49%, and 26% for patients with ASM, SM-AHN, and MCL, respectively.32 The estimated median OS for all patients with AdvSM was 46.9 months in a pooled analysis of patients from EXPLORER and PATHFINDER initiated with avapritinib ≤200 mg daily.47 Compared with a historical cohort of patients with AdvSM treated with best available therapy, avapritinib-treated patients exhibited significantly improved survival (adjusted hazard ratio, 0.48; 95% confidence interval, 0.29-0.79; P = .004), longer duration of treatment (23.8 vs 5.4 months; P < .001), and a 60% greater mean difference in the percent maximum decrease in levels of serum tryptase.51 An indirect treatment comparison found that avapritinib improved survival compared with midostaurin.52
Avapritinib: adverse events
In the pooled outcomes from the overall safety population of 131 avapritnib-treated patients,47 the most common nonhematologic adverse events (AEs) (all grades %/grade ≥3%) were peripheral/periorbital edema (81%/4%), diarrhea (34%/<1%), nausea (31%/3%), fatigue/asthenia (28%/7%), and cognitive effects (25%/2%).41 Hematologic AEs consisted of neutropenia (17%/16%), anemia (44%/27%), and the grouped terms thrombocytopenia/platelet count decreased (50%/30%). In EXPLORER, intracranial bleeding (ICB; eg, intraparenchymal hemorrhage, subdural hematoma) emerged as an AE of special interest that occurred in 9 (13%) patients, with 7 of these cases developing in the setting of thrombocytopenia (platelet count <50 × 109/L). Asymptomatic cases (n = 5, grade 1) were detected by prespecified protocol magnetic resonance imaging of the brain; 2 events were grade 2, and one each was grade 3 and grade 5 (associated with head trauma). Mitigation procedures (eg, exclusion of patients with a starting platelet count <50 × 109/L, dose hold and reduction for emergent thrombocytopenia reaching this threshold, platelet transfusions, increased blood count surveillance) led to a decrease in ICB in the interim PATHFINDER analysis, with only 1 patient (1.6%) experiencing a grade 2 subdural hematoma in the setting of progressive thrombocytopenia.45 The recommended frequency of platelet count monitoring is detailed in the avapritinib prescribing insert.
Emerging KIT inhibitors
BLU-263 and bezuclastinib
BLU-263 (Blueprint Medicines) has comparable selectivity and potency to avapritinib but limited brain penetration potential, which may mitigate cognitive changes and ICB. It is currently being evaluated in the phase 2/3 HARBOR study (NCT04910685) of patients with ISM.53 Substantial clinical interest exists in evaluating BLU-263 in patients with AdvSM as monotherapy or as sequenced therapy with AHN-directed agents given the concern about the potential for thrombocytopenia-emergent ICB in this patient population with avapritinib.
Bezuclastinib (CGT9486; Cogent Biosciences) is a type I TK inhibitor with activity against mutations in KIT exons 9, 11, 17, and 18, including D816V.54 Its target profile avoids other kinases such as WT KIT, PDGFRα, PDGFRβ, wild-type KIT, VEGFR2, and CSF1R and exhibits limited blood-brain barrier penetration and no central nervous system toxicities in preclinical studies. The drug is currently being evaluated in a phase 2, open-label clinical trial (APEX; NCT04996875) of AdvSM in which patients are first randomized to 1 of 4 doses of the drug in a dose optimization stage, followed by dose expansion at the recommended phase 2 dose. A phase 2/3 study of bezuclastinib in ISM/smoldering SM has also been initiated (NCT05186753).
AdvSM and KIT inhibitors: open questions and future directions
Avapritinib, midostaurin, or enrollment in clinical trials should be considered the first-line treatment option for AdvSM. While avapritinib is not recommend for patients with platelet count <50 × 109/L, it may otherwise merit preferred status. The drug can generate molecular remissions of KIT D816V, and recent data indicate more favorable long-term outcomes compared with historical treatments, including midostaurin. The major challenge of these KIT inhibitors is the AdvSM variant SM-AHN, in which prognosis is usually driven by the AHN, and the presence of a complex mutational landscape beyond KIT D816V may promote progression and resistance. In this regard, how to sequence KIT inhibition with AHN-targeted therapy is a major focus of clinical trial development in AdvSM. Table 2 highlights some of the major challenges and open questions that have emerged in the era of KIT inhibitors.
CLINICAL CASE (Continued)
The patient was started on avapritinib 200 mg daily. After 4 months, he became paracentesis independent. By 6 months, the albumin had increased to 4.8 g/dL, and the patient had gained 30 pounds and no longer exhibited muscle wasting. In addition, the serum tryptase level normalized to 10.1 ng/mL and the BM biopsy specimen showed no mast cell aggregates. He exhibited a 38% improvement in total symptom score on the AdvSM Symptom Assessment Form. Due to persistence of periorbital edema and achievement of a complete remission, the avapritinib dose was decreased to 100 mg daily. After 3 years of follow-up, he maintains a complete remission; the BM shows no evidence of MC aggregates and blasts are not increased. The serum tryptase level remains normal. The del(13q) abnormality and TET2 mutation persist, but the KIT D816V mutation is no longer detectable.
Primary eosinophilic neoplasms
The WHO major category of “myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1 or with PCM1-JAK2” has had its title changed to “myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions” in both the fifth edition of the WHO classification of hematolymphoid tumors and the International Consensus Classification (ICC).1,2 The modification in nomenclature reflects the common molecular theme of rearranged, constitutively activated tyrosine kinases. These TK gene fusions (and associated chromosomal breakpoints) involve PDGFRA (the most common fusion, FIP1L1::PDGRA, is cryptic; otherwise, breakpoint 4q12), PDGFRB (5q31~q33), FGFR1 (8p11), JAK2 (9p24), FLT3 (13q12), and ETV6::ABL1. ETV6::ABL1 presenting as Philadelphia chromosome-like acute lymphoblastic leukemia (which should be distinguished from this category of TK gene fusions) can also rarely present as de novo T-cell acute lymphoblastic leukemia or a myeloid neoplasm.55,56
Eosinophilia is not an invariable feature of these neoplasms but can serve as a useful pre-diagnostic checkpoint to think about these disease entities. At the time of writing, there have been 8 PDGFRA partner fusion genes characterized, over 30 for PDGFRB, 16 for FGFR1, 3 for JAK2, and 7 for FLT3 (Table 3).55-57 In some cases besides the cytogenetically invisible FIP1L1:: PDGFRA, standard cytogenetics and/or fluorescence in situ hybridization (FISH) may not be able to identify the TK gene fusion. Integrated genomic analyses, including chromosomal microarrays, whole-genome sequencing, and/or RNA sequencing, may be necessary to uncover small deletions or inversions resulting in cryptic fusion genes.58
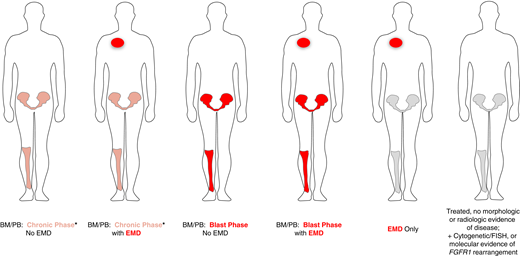
The clinical presentation of myeloid/lymphoid neoplasms with eosinophilia (MLN-eo) with TK gene fusions can be highly complex. One should determine whether the disease involves the BM and peripheral blood (PB) only, extramedullary disease (EMD) only, or both BM/PB and EMD. If clinically suspected, imaging (preferably positron emission tomography/computed tomography) should be undertaken to identify EMD so it can be serially followed during treatment. Second, the presence of chronic phase (CP) or blast phase (BP) disease should be characterized in the BM/PB. The presence of EMD represents a BP component. Last, the disease lineages of the BM/PB and EMD components should be characterized. The most common CP presentations of MLN include myeloid neoplasms: MPN or MDS/MPN (CMML or MDS/MPN-unclassifiable) with or without eosinophilia. The BM may exhibit an atypical (usually interstitial) infiltrate of MCs in the absence of the KIT D816V mutation. BP disease in the BM/PB and/or EMD may present as AML, B- or T-cell leukemia/lymphoblastic lymphoma, or a mixed-phenotype acute leukemia. Importantly, the disease lineage in the BM/PB can be different from the EMD; therefore, biopsy of the EMD for lineage ascertainment may be helpful but not always possible. In some cases, we have seen tandem involvement of the BM/PB by both a chronic myeloid neoplasm and B- or T-cell acute lymphoblastic leukemia. If patients have received prior treatment, only cytogenetic, FISH, or molecular evidence of a TK fusion gene may be evident as a marker to follow during subsequent therapy. The heterogeneous presentations of MLN with TK fusion genes are shown in Figure 2. Their heterogeneity may reflect the TK fusion gene, its partner, or the presence of additional cytogenetic or molecular abnormalities (eg, RUNX1 mutation in patients with FGFR1 fusions).
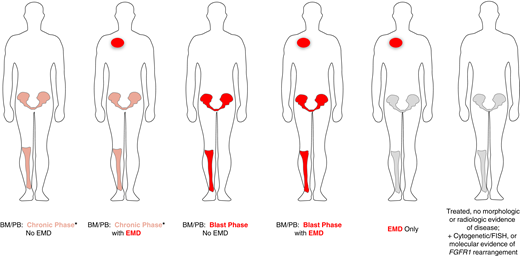
Clinical presentations of MLN with eosinophilia and tyrosine kinase gene fusions. *BM/PB can present with a concurrent chronic myeloid neoplasm as well as acute lymphoblastic leukemia/lymphoma (B- or T-cell lineage) or mixed-phenotype acute leukemia.
Clinical presentations of MLN with eosinophilia and tyrosine kinase gene fusions. *BM/PB can present with a concurrent chronic myeloid neoplasm as well as acute lymphoblastic leukemia/lymphoma (B- or T-cell lineage) or mixed-phenotype acute leukemia.
Imatinib in PDGFRA and PDGFRB TK gene fusions
FIP1L1::PDGFRA is the prototypic fusion TK gene and is almost always associated with eosinophilia. An increase in the vitamin B12 and/or serum tryptase level often accompanies the finding of atypical, interstitial MCs in the BM.59-61 Most patients are male who present with a chronic myeloid neoplasm with eosinophilia; however, rare cases of AML or T-cell ALL, as well as isolated or concurrent EMD, have been reported.62 FIP1L1:: PDGFRA results from a submicroscopic deletion of 800 kb on chromosome 4q12 and is not detected by standard karyotyping.59 FISH for the CHIC2 deletion63 or reverse transcription PCR is used to identify the fusion and for serial monitoring; false-negative cases using FISH have been reported and therefore PCR should be employed in such cases with high clinical suspicion.64
Studies have confirmed the deep and durable responses of FIP1L1::PDGFRA-positive disease to imatinib at starting doses of 100 mg daily.65-68 In patients with known or possible cardiac involvement with eosinophils, concomitant initiation of corticosteroids (eg, prednisone 1 mg/kg for 7-10 days) with imatinib is recommended to mitigate potential complications from heart failure/cardiogenic shock. Complete hematologic remissions are achievable in >95% of patients and can achieved within several weeks; complete FISH and/or PCR responses are typically observed within 3 to 6 months. In patients achieving FISH or PCR negativity, maintenance dosing of 100 mg 3 times a week or weekly can maintain deep responses.69 Similar to chronic myeloid leukemia, increasing experience is accumulating with imatinib discontinuation and the concept of treatment-free remission in FIP1L1::PDGFRA-positive cases. In case series, hematologic and/or molecular relapse has been documented in <6 months, but in some cases, molecular remissions have been maintained for more than 5 years.70-73 In a retrospective French series, the relapse-free survival rate of patients who discontinued imatinib was 57% after a median follow-up of 58 months (range, 25-100 months).74 Relapses occurred after a median of 10 months (range, 4-23), consisting of hematologic (n = 18, 90%) or isolated molecular relapses (n = 2, 10%). Imatinib was successfully resumed in 17 of 20 (85%) relapsed patients, with complete hematologic remission within 1 month in all cases and molecular responses within 12 months in all tested patients.74 In multivariable analysis, duration of prior imatinib therapy treatment was a statistically significant factor for relapse while time to imatinib initiation showed a trend for significance. After a mean follow-up of 80 months, the 1-, 5-, and 10-year overall survival rates of imatinib-treated patients in this cohort were 99%, 95%, and 84%, respectively.74
In another study, 12 patients with FIP1L1::PDGFRA who achieved a CMR were followed after imatinib discontinuation.75 Median time of treatment and median time of CMR before imatinib discontinuation were 80 months (range, 43-175 months) and 66 months (range, 37-174 months), respectively. A molecular relapse was observed in 4 patients between 10 and 24 months. A second CMR was achieved in 3 patients after 3, 4, and 21 months. Eight patients (62%) are in ongoing CMR (median, 17 months; range, 3-71 months). Molecular relapse-free survival was 91% at 12 months and 65% at 24 months.75 No significant differences were identified between patients with and without molecular relapse regarding dose and duration of imatinib therapy or prior duration of CMR. Further prospective studies are needed to better define predictive factors for treatment-free remission in these patients.
For patients with fusion tyrosine kinases involving PDGFRB, starting doses of imatinib 100 to 400 mg daily are recommended.55-57 A maintenance dose of 100 mg daily can be considered in patients achieving hematologic and molecular remissions following an induction dose of 400 mg daily. Long-term treatment outcomes with imatinib are similar to the experience with FIP1L1::PDGFRA.76 Long-term follow-up (median, 10.2 years) of PDGFRB-rearranged patients treated with imatinib for a median duration of 6.6 years showed a 96% response rate and a 10-year overall survival rate of 90%.77 None of the patients achieving a complete cytogenetic (n = 13) or molecular (n = 8) remission lost their response or exhibited progression to blast phase disease.77
Imatinib monotherapy can be effective in de novo presentations of blast phase disease with PDGFRA or PDGFRB fusion TK genes. In a case series of 17 patients in blast phase and/or with EMD, 15 patients treated with imatinib only achieved durable complete hematologic and molecular remissions.78 Two of 17 patients (12%) died after a median follow-up of 65 months. While these data are encouraging, an alternate approach may be to combine lineage-specific induction chemotherapy + imatinib with consideration of allografting patients in first complete remission.
Primary resistance is not a clinical concern. Although uncommon, secondary resistance almost always defaults to 2 canonical mutations: PDGFRA T674I or PDGFRA D842V. Although these mutations have exhibited variable sensitivity to tyrosine kinase inhibitors (TKIs) in vitro, clinical responses have been underwhelming.79-83 However, avapritinib's activity against PDGFRA D842V in patients with GIST42 augurs promise for similar benefit in the context of FIP1L1::PDGFRA.
JAK2, FLT3, and ETV6::ABL1 TK gene fusions
In several case series, patients have been treated with ruxolitinib for JAK2 gene fusions or with FLT3 inhibitors (midostaurin, sunitinib, sorafenib, gilteritinib) for gene fusions involving FLT3.84-86 Hematologic and cytogenetic remissions have been described with JAK2 or FLT3 inhibitors in both TK-driven neoplasms, but they tend not to be durable.79-85 Since JAK2- and FLT3-targeting TKIs in these diseases have generally not provided sufficient disease control, the intent should be to use them as a cytoreductive bridge to HSCT. This was recently demonstrated in an infant with ETV6::FLT3 MLN who achieved a morphologic, immunophenotypic, and cytogenetic remission with gilteritinib before proceeding to transplant.87 In patients with CP presentations of ETV6::ABL1-positive disease, the limited experience to date suggests that the second-generation TKIs nilotinib and dasatinib may elicit more durable complete remissions than imatinib.86
FGFR1 inhibitors in MLN with FGFR1 gene fusions
Patients with FGFR1-rearranged MLN-eo exhibit an aggressive disease course.88,89 Individuals with chronic phase disease exhibit a cumulative rate of transformation to blast phase at 12 months approaching 50%, and 1-year OS of patients presenting with blast phase disease is 30%.89 Hydroxyurea and multikinase inhibitors with anti-FGFR1 activity (eg, ponatinib, midostaurin) in CP and lineage-specific induction chemotherapy ± TKI in BP can lead to partial response (PR) or short-lived complete response (CR), but cytogenetic responses (CyR) are rare.90-92 Allogeneic HSCT is the only treatment modality that has been shown to produce long-term remissions in MLN with a FGFR1 fusion gene.93
Futibatinib is a selective inhibitor of FGFR1-4 that was evaluated in a patient with a PCM1::FGFR1 fusion.94 Futibatinib at a dose of 20 mg daily (dose reduced to 16 mg daily after 3 months for a grade 2 bullous rash) produced a durable, complete hematologic and cytogenetic remission that was ongoing after >18 months of therapy. Futibatinib is currently being evaluated in a phase 2 study of MLN as well as solid tumors (NCT04189445).
The most mature data are available for pemigatinib (INCB054828), a selective inhibitor of FGFR1-3. It has received regulatory approval for adult patients with previously treated, unresectable, locally advanced, or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangements.95 The ongoing FIGHT-203 study evaluating pemigatinib is a phase 2, multicenter trial enrolling adults with MLN-eo with a FGFR1 fusion TK gene. Initially, patients had to have received ≥1 prior therapy with a starting dose of pemigatinib 13.5 mg daily in 3-week cycles (2 weeks on, 1 week off). With amendments, patients without prior therapy were also eligible and the starting dose was modified to 13.5 mg daily on a continuous schedule. The primary end point is CR rate; secondary end points include overall response (ORR [CR + PR]), complete CyR or partial CyR, and safety.96 All primary and secondary end points were investigator assessed and also adjudicated retrospectively by a Central Review Committee (CRC) with CRC-defined criteria based on local lab and radiologic results and central review of histopathology and standard karyotyping/FISH.
Thirty-four patients were enrolled and treated (1 subject without an FGFR1 rearrangement was excluded from the efficacy analysis). The average number of prior therapies was 1.6 (range, 0-6); 3 patients had undergone prior transplant and 5 patients were treatment naive.96 The longest duration of pemigatinib exposure was 192 weeks with a median dosing duration of 29 weeks. Patients completed a median of 10.0 treatment cycles (range, 2-65). At data cutoff (December 31, 2020), treatment was ongoing in 18 patients (53%); reasons for treatment discontinuation (n, %) included bridging to transplant (n = 6, 18%), progressive disease (n = 5, 15%), adverse event (n = 3, 9%), physician decision (n = 1, 3%), and patient decision (n = 1, 3%).96
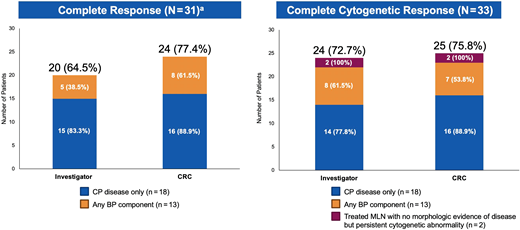
The baseline features of the 33 patients with FGFR1-rearranged MLN have been previously reported.96 Two patients had treated MLN with FGFR1 with a persistent cytogenetic abnormality only (no morphologic evidence of disease), and the remaining patients had CP (n = 18) or BP (n = 13) disease. Among these 31 patients, CR rates per investigator and CRC assessments were 64.5% and 77.4%, respectively; among the 33 patients evaluable for CyR, complete CyR rates were 72.7% and 75.8%, respectively (Figure 3).

FIGHT-203 Trial: Rates of complete clinical and cytogenetic response with pemigatinib. Responses to pemigatinib were adjudicated by local investigators and according to a central review committee. Central FISH was given priority over local cytogenetic results during CRC adjudications. aIn the 2 patients with treated MLN with no morphologic evidence of disease but persistent cytogenetic abnormality, cytogenetic responses were adjudicated, but clinical responses were not.
FIGHT-203 Trial: Rates of complete clinical and cytogenetic response with pemigatinib. Responses to pemigatinib were adjudicated by local investigators and according to a central review committee. Central FISH was given priority over local cytogenetic results during CRC adjudications. aIn the 2 patients with treated MLN with no morphologic evidence of disease but persistent cytogenetic abnormality, cytogenetic responses were adjudicated, but clinical responses were not.
The most common treatment-emergent adverse events (TEAEs) were hyperphosphatemia (68%), alopecia (59%), diarrhea (50%), stomatitis (44%), and anemia (35%). Grade ≥3 TEAEs in ≥10% of patients were anemia (18%) and pain in extremity and stomatitis (both 12%). Dose modifications due to any TEAE included dose interruption (65%), reduction (59%), and discontinuation (12%: due to cardiac failure, multiple-organ dysfunction, increased alkaline phosphatase, and calciphylaxis).96
While Kaplan-Meier median durations of CR and ORR had not been reached in the overall efficacy population, clinical and cytogenetic responses in BP disease were less frequent and less durable than in patients with CP disease. The most frequent reason for treatment discontinuation was bridging to transplant, which was achieved in 23% of BP patients.96 Taken together, these data indicate that pemigatinib can produce durable and high rates of complete clinical and cytogenetic responses, most of whom had progressed on prior therapy. Pemigatinib may be a useful option for patients ineligible for transplant or may facilitate bridging to transplant in eligible patients. Pemigatinib was approved by the FDA on August 26, 2022, for relapsed or refractory MLN with FGFR1 rearrangement.
Chronic eosinophilic leukemia
In both the new ICC and WHO classifications,1,2 diagnostic criteria for chronic eosinophilic leukemia (CEL) require both abnormal BM morphology (eg, dysplastic megakaryocytes with or without dysplastic features in other lineages or increased blasts ≥5% in the bone marrow and/or ≥2% in the peripheral blood) as well as demonstration of a clonal cytogenetic abnormality and/or somatic mutation(s). In the absence of a clonal cytogenetic abnormality and/or somatic mutation(s) or increased blasts, the aforementioned BM findings are sufficient in the presence of persistent eosinophilia, for which other causes have been excluded. The focus on abnormal BM histopathology97 is intended to distinguish CEL from idiopathic hypereosinophilic syndrome and hypereosinophilia of unknown significance. The ICC also requires persistent PB hypereosinophilia (eosinophil count ≥1.5 × 109/L and eosinophils ≥10% of white blood cells)1 while the WHO specifies the hypereosinophilia should endure for at least 4 weeks from the prior requirement of 6 months.2 The WHO also removed the CEL disease qualifier “NOS.”2 Notably, CEL is still included among the BCR::ABL1-negative MPNs, not MDS/MPNs, despite dysplastic marrow findings being a core histopathologic feature of these neoplasms.
CEL lacks recurrent cytogenetic and/or molecular genetic abnormalities such as BCR::ABL1 or TK gene fusions associated with MLN-eo. Case reports and series have annotated nonspecific abnormalities, including del(13q), del(20q), trisomy 8, monosomy 7, and complex karyotypes.98 In some CEL cases, next-generation sequencing (NGS) panels have uncovered gene mutations that are promiscuous among myeloid neoplasms (eg, DNMT3A, ASXL1, TET2, EZH2, SETBP1, CBL, SF3B1, among others).99 It may not always be possible to distinguish whether some variants are drivers of eosinophilia or clonal hematopoiesis of indeterminate potential mutations. Rarely, NGS may uncover potential druggable targets or pathways, such as the JAK-STAT signaling axis. In 1 study, the STAT5B N642H mutation was identified in 27 of 1715 (1.6%) cases referred for eosinophilia.100 Somatic insertion/deletion mutations (p.L583_A586delinsS) within exon 13 of the pseudokinase domain of JAK2 have also been described in patients with a diagnosis of both CEL and polycythemia vera,101 and a somatic activating JAK1 R629_S632delinsSA mutation was identified in another patient with long-standing CEL.102
CEL carries a poor prognosis, with a median OS in the range of 1 to 2 years in 2 case series.98,103 No consensus standard frontline treatment exists for CEL. Corticosteroids, hydroxyurea, PEG-IFN-α, and imatinib can improve leukocytosis and hypereosinophilia, but responses tend not to be durable. PEG-IFN-α can elicit hematologic and cytogenetic responses, as well as reversion of end-organ damage, including in patients who are relapsed/refractory to corticosteroids and hydroxyurea.55,56 In the absence of a tyrosine kinase target, hematologic improvement with empiric use of imatinib may reflect nonspecific myelosuppression, and responses tend to be short-lived. Anti–interleukin 5 and anti–interleukin 5 receptor antibodies (mepolizumab and benralizumab, respectively), which demonstrate activity in idiopathic hypereosinophilic syndrome and lymphocyte variant hypereosinophilia, are generally not active in primary eosinophilic neoplasms.104,105 As BM dysplasia is a prominent feature of bona fide CEL, the role of hypomethylating agents should be further explored in these cases.
Novel agents in primary eosinophilic neoplasms: open questions and future priorities
Imatinib has reversed the once poor survival of patients with PDGFRA and PDGFRB fusions into “good-risk” diseases. Despite the availability of TK inhibitors in MLN with rearranged FGFR1, JAK2, FLT3, and ABL1, responses to targeted agents are usually partial and less durable. Their natural histories generally remain poor and should be distinguished from the very favorable experience with imatinib in PDGFRA- and PDGFRB-rearranged disease. The use of pemigatinib as a bridging agent to transplantation in MLN with FGFR1 fusions exemplifies the approach that will need to be considered more broadly in MLN, especially in the BP of disease, where TKI response is often short-lived. For patients with CEL, the results of NGS may provide druggable targets in rare instances, but otherwise a treatment backbone with hypomethylating agents or PEG-IFN-α or enrollment in clinical trials of novel agents is recommended with an eye toward transplantation when feasible. Table 4 highlights outstanding questions and future priorities for the treatment of primary eosinophilic neoplasms.
Acknowledgments
Dr. Gotlib expresses gratitude to the Charles and Ann Johnson Foundation for its support of MPN Research at Stanford, as well the Stanford Hematology Division, US and international SM investigators, and patients with SM and their families. Dr. Gotlib expresses special acknowledgment of members of the Stanford mastocytosis clinical research collaborative: Dr. William Shomali, Dr. Asiri Ediriwickrema, Cheryl Langford, Justin Abuel, Cecelia Perkins, Lenn Fechter, Annie Jinsuh Jung, Denise DeVore, Leslie Hwang, Robert Jones, Parveen Abidi, and Bhuvana Ramachandran.
Conflict-of-interest disclosure
Research funding: Incyte, Novartis, Kartos, Blueprint Medicines, Cogent Biosciences, Abbvie, Celgene, BMS, Protagonist Therapeutics. Advisory boards/consulting/honoraria: Incyte, Novartis, Kartos, Blueprint Medicines, Cogent Biosciences, Abbvie, Protagonist Therapeutics, PharmaEssentia, Imago Biosciences.
Off-label drug use
Bezuclastinib and BLU-263 are under investigation for indolent and advanced SM. Futibatinib is under clinical investigation for FGFR1-rearranged MLN. Off-label use: ruxolitinib, fedratinib, pacritinib, and momelotinib for JAK2-rearranged MLN; midostaurin and ponatinib for FGFR1-rearranged MLN; midostaurin, sorafenib, sunitinib, and gilteritinib for FLT3-rearranged MLN; and imatinib, dasatinib, nilotinib, bosutinib, ponatinib, and asciminib for ETV6::ABL1-rearrranged MLN.