
Abstract
Recent years have brought an explosion of new diagnostic tools to the pathology of lymphomas, which have permitted more precise disease definition and recognition of factors that can predict prognosis and response to treatment. These new methods exploit both the biological features of normal lymphocytes as they progress through differentiation pathways and the genetic abnormalities that characterize malignant transformation. These features can be assessed in individual tumors with techniques that detect proteins (immunophenotyping), messenger RNA (in-situ hybridization), or changes in DNA [Southern blot, PCR, fluorescence in-situ hybridization (FISH), and gene sequencing]. Recently, the novel technology of “gene chips” or DNA microarrays has greatly enhanced the efficiency of analyzing expression of many genes simultaneously at the RNA level. Understanding the relationship of lymphoid neoplasms to their normal counterparts and the genetic events that lead to malignant transformation in lymphoid cells are essential for physicians caring for patients with lymphoma, since these are the basis of modern classification, diagnosis, and prognosis prediction. Although microarray technology is not ready for prime time in the daily diagnosis of lymphoma, practitioners should understand its potential and limitations.
The vast majority of lymphoid neoplasms worldwide are derived from B lymphocytes at various stages of differentiation. The review by Harald Stein and colleagues present the events of normal B-cell differentiation that are relevant to understanding the biology of B-cell neoplasia. These include antigen receptor [immunoglobulin (Ig)] gene rearrangement, somatic mutations of the Ig variable region genes, receptor editing, Ig heavy chain class switch, and differential expression of a variety of adhesion molecules and receptor proteins as the cell progresses from a precursor B cell to a mature plasma cell. Most lymphoid neoplasms have genetic abnormalities, many of which appear to occur during the gene rearrangements and mutations that characterize normal B-cell differentiation. Dr. Riccardo Dalla Favera reviews the mechanisms of these translocations and other abnormalities, and their consequences for lymphocyte biology. The association of specific abnormalities with individual lymphomas is reviewed. Dr. Wing C. Chan reviews the technology and applications of DNA microarray analysis, its promises and pitfalls, and what it has already told us about the biology of lymphomas. Finally, what does this all mean? The applications, both current and future, of these discoveries to the diagnosis and treatment of patients with lymphoma are discussed by Dr. Nancy Lee Harris.
I. Genetic Events and Gene Expression in B-Cell Differentiation: Implications for Lymphoma Classification
Harald Stein, MD,*
Institut für Pathologie, Universitätsklinikum Benjamin Franklin, Freie Universität Berlin, Hindenburgdamm 30, D12200 Berlin, Germany
A prerequisite for an understanding of B-cell lymphomas and their classification is the knowledge of the structure, cellular composition, changes in gene expression and molecular events involved in the differentiation and function of normal B-cells. This review will provide information that helps the understanding of B-cell neoplasms in relation to the differentiation events of the normal B-cell system.
Structure of the lymphoid system
Cellular composition of the lymphoid tissues
Lymphoid tissue, together with recirculating lymphocytes, constitutes the lymphoid system, which serves as one of the defence mechanisms of the organism against bacteria, viruses, parasites and toxins. The following cells, which are all involved in the defence and/or in the regulation of immune response, can be identified in lymphoid tissue:
B cells
T cells
Natural killer (NK) cells
Macrophages
Follicular dendritic cells (FDC)
Interdigitating dendritic cells (IDC)
High endothelial venules (HEV)
Organization of the lymphoid tissue
Two major forms of lymphoid tissue have been distinguished: central (primary) lymphoid tissue and peripheral (secondary) lymphoid tissue
Central lymphoid tissue is composed of the bone marrow and the thymus. The bone marrow is where B cells primarily arise and differentiate to mature B cells, and the thymus is where premature T cells differentiate into mature T cells. Mature B and T cells express antigen receptors, each with a different antigen specificity. These mature B and T cells migrate into the peripheral lymphoid tissues, which consist of blood, the spleen, lymph nodes and the mucosa associated lymphoid tissue (MALT). They steadily recirculate throughout the body so that there is a high probability that they will meet any foreign antigen penetrating the body. If such an encounter takes place, effector cells and memory cells evolve.
Differentiation of B cells in the central and the peripheral lymphoid tissues involves changes in cytology and homing, which are correlated to genetic events and changes in gene expression.
B-Cell Differentiation
Precursor B cells
B cells develop from hematopoietic stem cells usually located in the bone marrow (Figure 1 ).1– 3 The first precursor B-cell identified is the progenitor B cell or pro-B cell. The pro-B cell expresses some B-cell characteristic antigens (Table 1 ) and initiates rearrangement of the immunoglobulin (Ig) gene locus. In Table 2 , the individual steps of Ig gene rearrangement are described, using the Ig heavy chain gene (IgH) as an example. The next precursor B cell stage is the pre-B cell. In this cell, the recombination of VDJ genes of the heavy chain gene locus is already complete, resulting in cytoplasmic expression of the μ heavy chain. The subsequent rearrangement of the Ig light chain gene (IgL) locus leads to the expression of a complete IgM molecule consisting of two μ-chains and two light chains (Figure 2; color page 544), which is expressed on the cell surface and serves as its receptor for antigen. This third precursor B cell stage is designated immature B cell. Immature B cells give rise to mature naïve B cells that, as a result of an alternative splicing of IgH mRNA, express both IgM and IgD. In contrast to immature B cells, mature (naïve) B cells have the capacity of responding to the binding of a foreign antigen. They respond by proliferating and differentiating into plasma cells and memory B cells. They are called naïve because they have not yet encountered an antigen to which their surface Ig receptor molecules are able to specifically bind.
Specific marker molecules for early developmental precursor stage of differentiation are CD34 and TdT (terminal desoxyribonucleotidyl transferase). Since both markers are not lineage specific, they have to be applied in conjunction with T- and B-cell markers in order to determine the cellular origin of the precursor cell disease. TdT has an important function, as it inserts N-segments between the V (variable) region, D (diversity) region and J (joining) region during the recombination of these region genes (Figure 2, Table 1). When the recombination process is complete, the TdT gene switches off, with the result that all post-precursor (peripheral or mature) B cells are TdT-negative.
Peripheral (mature) B cells
Mature naïve B cells.
Mature naïve B cells populate the blood as recirculating IgM+D+ small B cells and the peripheral lymphoid organs, where they form primary B-cell follicles in association with FDC. There is evidence to suggest that these mature (post-bone marrow) naïve B cells are not homogenous, but probably consist of three subsets:
Recirculating subset expressing CD23 and non-auto-antigen-reactive Ig receptors
Recirculating subset expressing CD23 and low affinity auto-reactive Ig receptors, also known as B1 cells
sessile naïve B cells lacking CD23 and expressing non-auto-antigen-reactive Ig receptors
Upon antigen encounter, mature naïve B cells appear to move into the T-cell zone of lymphoid tissues, where they transform into large B-blasts and then proliferate. The daughter cells either differentiate into short-lived, IgM-producing plasma cells or into B cells that acquire the capacity to initiate a germinal center reaction. These so-called primed B cells move into primary follicles, where they proliferate and differentiate into centroblasts to form an early germinal center. The non-antigen-triggered naïve B cells of the primary follicle are pushed aside and, thus, form the follicle mantle, or mantle zone (Figure 1). This follicle, containing a germinal center and a mantle, is known as a secondary follicle.
The Germinal Center Reaction.4– 7
This process serves the functions listed in Table 3 . These functions require differentiation steps, changes in gene expression, and the introduction of mutations in the rearranged Ig variable (IgV) region genes.
Germinal center B cells.
Early germinal center B cells are rapidly proliferating B cell blasts, which are usually called centroblasts. These centroblasts differentiate further into centrocytes, which gather at one end of the follicle. This leads to the formation of two zones within the germinal center: a “dark zone” composed of centroblasts and a “light zone” containing mainly centrocytes. The centroblasts and centrocytes differ from mature naïve B cells in their immunophenotype and sensitivity to apoptosis: (i) they express CD10 and BCL6; (ii) they up-regulate the expression of the transcription factors Oct2 and BOB.1; (iii) they down-regulate the expression of surface Ig receptors; and (iv) they down-regulate the expression of the anti-apoptosis protein BCL2. Apoptosis of the germinal center B cells can be prevented only by survival signals delivered by FDC and T cells.
Follicular dendritic cells.
The formation of germinal centers is linked to an increase in FDC, whose slender cell processes form a dense network that is particularly high in the light zone. The cellular processes of the FDC cannot be recognized in conventional stains, and their visualization requires immunostaining with CD21 antibodies (which are directed at the complement receptor C3d) or CD35 antibodies (which are directed against the complement receptor C3b). The FDC of the light zone also express CD23.
A major task of FDC is the trapping and presentation of unprocessed antigen, which serves in the selection of germinal center B cells by antigen. As described above, the antigen provoking the immune response first leads to the generation of short-lived plasma cells that produce low-affinity antibodies of IgM type. These IgM antibodies bind to the antigen and form antigen-antibody complexes that activate complement and bind C3b and C3d. The antigen-antibody-complement complexes are captured by the FDC via their C3b and C3d receptors and, thus, are presented to the germinal center B cells.
Antigen selection and hypermutation (affinity maturation)8– 10
The major goal of the germinal center reaction is the generation of B cells that produce immunoglobulin with high affinity for the antigen(s) that provoked the immune reaction. This goal is achieved by the rapid proliferation of B cells, a randomized introduction of mutations into the Ig receptor gene region that codes for the antigen binding site, and subsequent elimination of those B cells that do not have high-affinity surface Ig receptors (Figure 3, color page 544). To increase the affinity of the Ig receptors on the B cells that are multiplied in the germinal center reaction, mutations are inserted into the IgV genes. Since this process is partially random, most of these mutations either do not increase, or even decrease, the affinity of the Ig receptors or even completely prevents the expression of Ig by creating stop codons or frame shifts. The centrocytes with these unfavorable mutations do not bind with high affinity to the antigen trapped on the FDC processes and do not receive survival signals. In fact, more than 90% of the centrocytes die as a result of apoptosis. These cells are phagocytosed and digested by the so-called starry sky macrophages. This selection process is made more efficient by the decrease of Ig receptor molecules expressed on the surface of the germinal center B cells. The few centrocytes whose mutations have resulted in surface Ig receptors with high affinity for the antigen presented by the FDC will bind to the trapped antigen and receive survival signals from the FDC (positive selection).
The presence of IgV region mutations is now considered a reliable marker for a cell that has been exposed to the germinal center—either a germinal center or post-germinal center stage. The presence of ongoing mutations—variations in the mutation pattern among germinal center B-cell clones—is characteristic of cells still at the germinal center stage.
Receptor editing.11
The specificity of the Ig receptors of the germinal center B cells may be modified by a further process designated receptor editing. In this process, the originally rearranged Ig gene segment, usually IgL, is replaced by another VL segment (Figure 4, color page 544).12
Class switch.
The affinity maturation process is associated with a switch of the Ig heavy chain class from IgM to IgG, IgA or less commonly to IgE. The “class-switched” B cells are those that preferentially mature into plasma cells.
Post-germinal center B cells
The centrocytes that survive in the germinal center mature into long-lived class-switched plasma cells or memory B cells.
Plasma cells.
Plasma cellular differentiation starts in the light zone of the germinal center. This can be evidenced by immunostains for the plasma cell-related marker VS38c and the nuclear transcription factor MUM1/IRF4. The plasmacellular differentiation is completed following emigration of the B-cells out of the germinal center. The long-lived plasma cells predominantly populate the bone marrow and organs that are directly exposed to foreign antigens, i.e. the gastrointestinal tract and the lung.
The plasmacellular differentiation is associated with both a loss and gain of molecules. The molecules that are lost or down-regulated are B cell antigens (e.g. CD19, CD20, CD22, BSAP) and surface Ig. The molecules that are gained are the transcription factor MUM1/IRF4, the rough endoplasmatic reticulum associated antigen VS38c, and the adhesion molecule CD138, and CD38. A functionally very significant change of plasmacellular differentiation concerns Ig production. The synthesis of surface Ig, i.e. the antigen receptors, is down-regulated and the production of Ig destined for secretion is augmented instead. The secretory Ig accumulates in large quantities in the cytoplasm where it is easily detectable by immunohistochemistry. The change in the destination of the Ig molecules is mediated by a change of “address sequences” attached to the synthesized Ig protein.
Memory B cells (marginal zone B cells):
It is not yet clear whether memory B-cell differentiation also begins within germinal centers. This lack of knowledge is due to non-availability of marker molecules that selectively stain this B-cell subset. The currently known features of memory B cells that may help distinguish them from other B-cell populations are:
strong expression of IgM
no or little expression of IgD
expression of CD2713
somatic mutations within IgV genes without signs of ongoing mutations
preferential homing to marginal zones
The latter feature prompted the designation marginal zone B cells. Well-developed, i.e. easily recognizable, marginal zones are usually only seen in the spleen and mesenteric lymph nodes, as well as in MALT. On the basis of FACS (fluorescence activated cell sorting) studies, immunologists distinguish IgM+ memory B cells and IgG+ memory B cells. The IgG+ memory B cells are not detectable in tissue sections using the immunohistochemistry methods currently available.
Relationship of B-cell Neoplasms to Physiological B Cell Subsets and Differentiation Stages14,15
Most B-cell neoplasms mirror the features of the different B-cell differentiation stages. This is especially true of small and medium-sized cell B-cell lymphomas.
Lymphoblastic leukemia/lymphomas resemble—in morphology and immunophenotype—(TdT+, CD34+CD10+) precursor B cells.
Around 50% of chronic lymphocytic leukemia of B-cell type (B-CLL) cases have features of activated mature naïve (non-mutated) B cells and the other 50% show characteristics of memory (mutated) B cells. The non-mutated B-CLL type may be related to the recirculating naïve B-cell subset also known as B1 cells, which express CD23 and low affinity auto-reactive surface Ig receptors.
Mantle cell lymphoma cells resemble primary follicle cells or follicle mantle cells in immunophenotype, in homing and in the presence of a loose network of FDC, i.e. the cells in the mantle zone that lack CD23 and probably correspond to sessile naïve B cells. They also resemble mantle cells in their IgV gene mutation pattern, since their IgV genes are either not mutated (∼ 90% of cases) or carry only a very few mutations.
Follicular lymphomas are similar to reactive secondary follicles in their cellular composition (centroblasts and centrocytes), their follicular arrangement with the formation of a dense FDC network, their expression of CD10 and BCL6 and increased expression of Oct2 and BOB.1. Genetically, they carry mutations in their IgV genes and show signs of ongoing mutations (intraclonal heterogeneity), characteristic of germinal center B cells.
Marginal zone lymphomas resemble physiological marginal zone cells in that they preferentially expand in marginal zones and tissues containing epithelial cells such as MALT, lung, salivary glands, etc. Further, they express IgM in the complete or partial absence of IgD. They typically have mutated IgV region genes without ongoing mutations.
Plasmacytoma/plasma cell myelomas are usually very similar in morphology and immunophenotype to non-neoplastic plasma cells. They usually lack CD45, CD20, and surface Ig and they express MUM1/IRF4, VS38c, CD138, CD38 and secretory Ig in the cytoplasm. Immature and anaplastic plasmacytomas may lack some of these markers.
Lymphocyte predominant Hodgkin lymphoma is a B-cell neoplasm with features of a germinal center cell origin. The neoplastic cells express CD45 and most B-cell antigens, have rearranged Ig genes and show somatic mutations characteristic of germinal center cells.
Classical Hodgkin lymphoma, in contrast, is a B-cell neoplasm that has lost nearly all morphological and immunophenotypical features of its cell of origin.15,16 Its derivation from germinal center B cells could only be demonstrated by genetic studies, showing that the neoplastic cells have rearranged Ig genes that are typically mutated, consistent with exposure to the germinal center. Despite its B-cell origin, the tumor cells of Hodgkin lymphoma lack most molecules that are characteristic of B cells and germinal center B cells, and have instead acquired molecules that are typically absent from germinal center B cells, including CD30, CD15, TARC, and TRAF1.
Diffuse large B-cell lymphomas stand—in terms of similarity to their cell of origin—between the small B-cell lymphomas and classical Hodgkin lymphoma. They display immunophenotypic and genetic features of B cells and B cell subsets, but often so incompletely that their precise allocation to a certain B-cell population is impossible. This lack of knowledge may soon be closed by cDNA micro-array studies, which will lead to the discovery of new genes characteristic of, or even specific to, certain B-cell subsets.
II. Molecular Pathogenesis of Non-Hodgkin's Lymphomas
Laura Pasqualucci, MD, and Riccardo Dalla-Favera, MD*
Institute of Cancer Genetics, Columbia University, 1150 St. Nicholas Avenue, Room 303, New York NY 10032
Non-Hodgkin's lymphomas (NHL) represent a heterogeneous group of diseases deriving from mature B cells (85% of cases) and, in a minority of cases, from T cells. Among B-NHL, most histologic subtypes arise from germinal center (GC) or post-GC B cells, since they have undergone hypermutation of the immunoglobulin variable region (IgV) genes, a phenomenon restricted to GC B cells (Figure 5 ).1
Analogous to most human cancers, the genetic lesions involved in NHL include the activation of proto-oncogenes and the disruption of tumor suppressor genes2. In contrast to many types of epithelial cancers, the genome of lymphoma cells is relatively stable and is not subject to the generalized random instability that characterizes many types of epithelial cancers.3 In addition, lymphomas generally lack microsatellite instability, which is caused by defects in DNA mismatch repair genes in some hereditary cancer predisposition syndromes as well as in a fraction of sporadic solid cancers.4,5 Historically, detection of recurrent, non-random chromosomal abnormalities by karyotypic analysis of NHL metaphases has represented the major clue toward the identification and cloning of most genetic alterations of NHL.
Activation of Proto-oncogenes by Chromosomal Translocation
Chromosomal translocation represents the main mechanism of proto-oncogene activation in NHL. Analogous to most types of hematopoietic neoplasms, chromosomal translocations in NHL represent reciprocal and balanced recombination events between two specific chromosomal sites. These translocations are characterized by recurrency within a specific clinico-pathologic category of NHL (Figure 5) and are clonally represented in each tumor case. All NHL chromosomal translocations that have been cloned until now share a common feature, i.e. the presence of a proto-oncogene mapping to the vicinity of one of the two chromosomal recombination sites. In contrast with neoplasms of precursor lymphoid cells, chromosomal translocations associated with mature B- and T-cell malignancies do not generally lead to coding fusions between two genes. Rather, they juxtapose the proto-oncogene to heterologous regulatory sequences derived from the partner chromosome (Figure 6 ). These sequences may derive from antigen receptor loci as well as from other loci that are expressed at sustained levels in normal cells corresponding to the differentiation stage of the lymphoma (Table 4 ). The common consequence of the translocation is the deregulated expression of a proto-oncogene by two mechanisms: homotopic deregulation and heterotopic deregulation. Homotopic deregulation occurs when the proto-oncogene is expressed in normal cells of the same tissue, but its regulation is changed in the tumor. Heterotopic deregulation occurs when the proto-oncogene is not physiologically expressed in the normal cells and becomes ectopically expressed as a consequence of the translocation. The two exceptions to the deregulation model of NHL translocations are represented by the t(2;5) of T-cell anaplastic large cell lymphoma and the t(11;18) of MALT lymphoma, which cause gene fusions coding for chimeric proteins (Table 4).6,7
The pathogenetic role of chromosomal translocations is demonstrated by in vitro transformation studies as well as by experiments in transgenic animal models. These experimental models indicate that chromosomal translocations contribute to lymphoma but are not sufficient to cause it, consistent with the requirement for multiple genetic lesions in tumorigenesis. The mechanism by which chromosomal translocations occur is largely unknown, although they appear to be associated with dysfunctions of the genetic remodeling mechanisms operating in lymphoid cells, including Ig gene rearrangements (VDJ and switch recombination) and somatic hypermutation.8
Inactivation of tumor suppressor loci.
Disruption of tumor suppressor loci in NHL occurs through mechanisms similar to those associated with other human cancers and generally leads to biallelic inactivation, most frequently through deletion of one allele and mutation of the other. The tumor suppressor genes most frequently involved in the pathogenesis of NHL are represented by p53, p16, and ATM (for ataxia telangiectasia mutated).9– 11
Somatic hypermutation.
Recent evidence suggests that important genetic changes associated with lymphomagenesis may derive from an apparently aberrant activity of the somatic hypermutation process that normally engenders Ig diversity in germinal center B cells by mutating the IgV genes.1,15 Somatic hypermutation may contribute to NHL development by three mechanisms. First, based on the observation that somatic hypermutation requires DNA double strand breaks,16 it has been suggested that it may favor the occurrence of chromosomal translocations. Second, it has been recently shown that the physiologic activity of somatic hypermutation is not restricted to IgV genes, since the 5′ sequences of the BCL-6 and Fas/CD95 genes are also hypermutated in normal GC B-lymphocytes.17–,19 Initial evidence suggests that some of these mutations may be selected during lymphomagenesis for their activity in deregulating BCL-6 gene expression (Pasqualucci et al, in preparation). Finally, recent evidence suggests that an apparently aberrant activity of somatic hypermutation can target multiple genes, including several proto-oncogenes, in DLBCL (see below).20
Pathogenetic Heterogeneity of NHL
Small lymphocytic lymphoma/B-cell chronic lymphocytic leukemia.
The molecular pathogenesis of small lymphocytic lymphoma/B-cell chronic lymphocytic leukemia (SLL/B-CLL) is largely unknown. In particular, none among the cancer-related genes known to date has been shown to associate consistently and selectively with SLL/B-CLL. Deletions of chromosome 13q14 occur in approximately 60% of cases when analyzed by sensitive molecular tools, but the tumor suppressor gene presumably involved in these lesions has not been identified.14 Among known cancer related genes, mutations of p53 occur in 10% of the cases of SLL/B-CLL and the frequency of p53 inactivation increases substantially in late stages of the disease, suggesting that it may be involved in tumor progression.21,22
Lymphoplasmacytic lymphoma.
Approximately 50% of lymphoplasmacytic lymphoma associate with the t(9;14)(p13;q32) translocation (Table 4 and Fig. 5).23 The translocation appears to display a preferential clustering with cases associated with Waldenström's macroglobulinemia. The chromosomal breakpoints of t(9;14)(p13;q32) involve the Ig heavy chain (IgH) locus on chromosome 14q32, and, on chromosome 9p13, a genomic region containing the PAX-5 (Paired Homeobox-5) gene.24PAX-5 encodes a B-cell specific transcription factor involved in the control of B-cell proliferation and differentiation.25 Presumably, the juxtaposition of PAX-5 to the IgH locus in NHL carrying t(9;14)(p13;q32) causes its deregulated expression, thus contributing to tumor development.
Mantle cell lymphoma.
Mantle cell lymphoma (MCL) is frequently associated with t(11;14)(q13;q32) (Table 4 and Fig. 5).26 The translocation juxtaposes the BCL-1 locus at 11q13 with the IgH locus at 14q32, leading to heterotopic deregulation of BCL-1 (also known as CCND1 or PRAD1), which encodes for cyclin D1, a member of the D-type G1 cyclins involved in cell cycle control26 and not expressed in normal B cells. BCL-1 is expressed and detectable by immunohistochemical analysis even in MCL cases lacking a cytogenetically detectable t(11;14)(q13;q32), strongly suggesting that deregulation of this gene is a critical event in the pathogenesis of MCL. The pathogenetic role of BCL-1 activation in human neoplasia is suggested by the ability of cyclin D1 deregulation to contribute to B-cell lymphomagenesis in transgenic mice.27,28 Among B-NHL, cyclin D1 overexpression is restricted to MCL and represents a useful diagnostic marker for this malignancy.
Follicular lymphoma.
Chromosomal translocations involving the BCL-2 gene.
Translocations involving 18q21 [t(14;18)(q32;q21)] typically juxtapose 18q21 to the IgH locus, leading to deregulated expression of BCL-2 and, consequently, to constitutively high levels of the BCL-2 protein within the cells.30 The BCL-2 gene encodes a 26-kDa integral membrane protein that has been localized mainly to mitochondria.31 Whereas most proto-oncogenes of lymphoid neoplasia directly enhance cell proliferation, BCL-2 controls the cellular apoptotic threshold by preventing programmed cell death.31 Thus, deregulation of BCL-2 expression may lead to the abnormal survival of B cells with accumulation of additional genetic lesions leading to lymphomagenesis.
Other genetic lesions.
Deletions of chromosome 6 at 6q27 occur in approximately 20% of the cases.12 Over time, a significant fraction of FL evolves into an aggressive lymphoma with a diffuse large cell architecture. This histologic transformation is frequently associated with p53 mutations/deletions.32 In some cases, transformation is accompanied by inactivation of p16 by deletion, mutation or hypermethylation. In very rare cases, the histologic progression of FL involves c-MYC rearrangements or chromosome 6q deletions.12
Mucosa-associated lymphoid tissue (MALT) lymphoma.
The understanding of the molecular pathogenesis of MALT lymphoma is still in its early stages. In the case of gastric MALT lymphoma, the majority of tumors are associated with Helicobacter pylori infection.33 It has been suggested that gastric MALT-NHL may be dependent upon antigen stimulation by H. pylori since malignant lymphoid cells respond to H. pylori antigens and since the lymphoma may regress upon eradication of infection. The most common genetic alteration of MALT-NHL is represented by the t(11;18)(q21;21) translocation (∼50% of cases), which fuses the API2 gene, encoding an inhibitor of apoptosis, to the MLT gene, generating a novel fusion protein with presumed anti-apoptotic functions (Table 4 and Fig. 5).7 More rarely, the t(1;14)(p22;q32) translocation leads to transcriptional deregulation of the BCL-10 gene, a negative regulator of apoptosis.34 Among genetic alterations commonly involved in other NHL types, only BCL-6 rearrangements and p53 mutations have been detected in MALT-NHL, though at very low frequency. Cytogenetic studies, however, have pointed to several abnormalities recurrently involved in these tumors, including trisomy 3.2
Diffuse large B-cell lymphoma (DLBCL).
DLBCL is characterized by a marked heterogeneity in phenotype and clinical behavior, suggesting that it may include multiple, presently unrecognized disease entities.29 Consistent with this heterogeneity, the genetic lesions associated with DLBCL are also heterogeneous.
Chromosomal translocations and mutations of BCL-6.
Cytogenetic studies have demonstrated that chromosomal alterations affecting band 3q27 are a recurrent abnormality in DLBCL (Table 4 and Fig. 5). These alterations are predominantly represented by reciprocal translocations between the 3q27 region and various (>10) alternative partner chromosomes, including, the sites of the Ig genes at 14q32 (IgH), 2p11 (Igk) and 22q11 (Igλ).35
The cloning of the 3q27 chromosomal breakpoints led to identification of the BCL-6 gene, which is involved in the majority of DLBCL cases harboring 3q27 breaks irrespective of the partner chromosome participating in the translocation.36 BCL-6 is a transcriptional repressor belonging to the family of transcription factors containing zinc-fingers, and functions by inhibiting the expression of genes carrying its specific DNA-binding motif.37
Within the B cell lineage, BCL-6 expression is topographically restricted to the GC, and is required for GC formation, since mice lacking BCL-6 consistently fail to form GC and display impairments in the T-cell dependent immune response.38,39 Overall, these animal models unequivocally demonstrate that BCL-6 is a key regulator of GC formation and B-cell immune response.
Chromosomal translocations involving band 3q27 are detectable in 35% of DLBCL cases and in a small fraction of FL.35 In these translocations, the BCL-6 gene is truncated in its 5′ non-coding region and heterologous promoters derived from other chromosomes are juxtaposed in front to an intact BCL-6 coding sequence, leading to homotopic BCL-6 deregulation by a mechanism called promoter substitution (Figure 7 ).40 In addition, up to 75% of DLBCL display multiple somatic mutations clustering in the BCL-6 5′ regulatory sequences independent of chromosomal translocations,17,41 suggesting that some mutations may be selected for their ability to alter its transcriptional regulation (Fig. 7).
Aberrant somatic hypermutation.
Recent findings indicate that an apparently aberrant activity of the somatic hypermutation mechanism, which normally targets the Ig, BCL-6 and Fas genes, can elicit tumor-associated lesions at multiple genetic loci in DLBCL.20 The four proto-oncogenes PIM-1, cMYC, PAX-5 and RhoH/TTF were found hypermutated in DLBCL, with > 50% of cases carrying at least two mutated genes. Mutations are of somatic origin, independent of chromosomal translocation to the Ig loci, and share features specific for the IgV-associated somatic hypermutation mechanism. Moreover, in PIM-1 and c-MYC the mutations affect non-translated as well as coding regions, leading to amino acid changes with potential functional consequences. In contrast with IgV, however, none of these four genes displayed a significant level of mutations in normal GC B-cells or in other GC-derived lymphomas, indicating a tumor-specific malfunction of somatic hypermutation in DLBCL. Intriguingly, each of the four hypermutable genes is also susceptible to chromosomal translocations in the same region, consistent with a role of hypermutation in generating translocations via DNA double-strand breaks.16 The number of genes targeted by the aberrant somatic hypermutation mechanism and the mechanism involved in this aberration are presently unknown. However, by mutating the regulatory and coding sequences of multiple genes and possibly by favoring chromosomal translocations, aberrant hypermutation may represent a major contributor to DLBCL development.
Other genetic lesions of DLBCL.
Several additional genetic lesions have been detected in DLBCL. Approximately 25% of DLBCL cases display chromosomal rearrangements of BCL-2.2 These translocations are entirely similar to the ones associated with FL and lead to the deregulated expression of BCL-2. These alterations appear to be mutually exclusive with BCL-6 rearrangements and tend to associate with DLBCL cases deriving from the histologic transformation of FL.42 Amplification of the REL gene, encoding a member of the NF-κB/REL family of transcription factors involved in cell activation and survival, occurs in 20% DLBCL, preferentially in cases with extranodal involvement.43 Among tumor suppressor genes, inactivation of p53 frequently associates with cases resulting from the histologic transformation of FL (Fig. 5). Finally, deletions of the long arm of chromosome 6 are also frequently detected in DLBCL, although the gene(s) involved is not known.13
Burkitt lymphoma: Translocations involving c-MYC.
The sporadic, endemic and HIV-associated forms of Burkitt lymphoma (BL) are characterized by chromosomal translocations between c-MYC and one of the Ig loci in 100% of cases (Table 4 and Fig. 5).2 The common consequence of these translocations is the homotopic deregulation of the c-MYC proto-oncogene, which encodes a ubiquitously expressed nuclear phosphoprotein that functions as a transcriptional regulator controlling cell growth and proliferation.44 Expression of c-MYC is rapidly induced in quiescent cells upon mitogenic induction, suggesting that c-MYC plays a role in mediating the transition from quiescence to proliferation. Chromosomal translocations cause c-MYC deregulation by at least two distinct mechanisms. First, translocated c-MYC alleles are juxtaposed to heterologous regulatory elements derived from Ig loci. Second, the 5′ regulatory regions of c-MYC are frequently affected by mutations, which in some cases have been shown to alter c-Myc transcription by releasing a block on transcriptional elongation.45 In other cases, missense mutations can deregulate c-Myc function by interfering with its phosphorylation, protein stability or repression of transactivation activity by the Rb-related protein p107.44
Several lines of experimental evidence document that deregulated expression of c-MYC can influence the growth of B-cells in vitro and in vivo. In particular, the targeted expression of c-MYC oncogenes in the B-cell lineage of transgenic mice leads to the development of B-cell malignancies.
Other genetic lesions of BL.
2 In addition to c-MYC translocations, the molecular pathogenesis of BL involves infection of the tumor clone by EBV, inactivation of the p53 and p16 tumor suppressor genes, mutations of the 5′ non-coding regions of BCL-6, and deletions of 6q.2 Infection by EBV occurs in virtually all cases of endemic BL and in approximately 30% of cases of sporadic BL.46 The consistent monoclonality of EBV infection in BL suggests that infection precedes clonal expansion of the tumor, consistent with a pathogenetic role of the virus. Notably, however, BL cells fail to express the EBV transforming antigens LMP-1 and EBNA-2, rendering the role of EBV infection unclear. Inactivation of p53 is detected in approximately 30-40% of BL cases, independent of their geographic origin or of the presence of EBV infection.22 Inactivation of p16 occurs in 30-40% of BL through mutation, deletion or hypermethylation.47 As in many other NHL types, BL is also associated with deletions of 6q.
Anaplastic large cell lymphoma.
Anaplastic large cell lymphoma (ALCL) is a T-cell lymphoma and typically associates with the t(2;5)(p23;q35) translocation, which involves the fusion of the nuclephosmin/B23 (NPM) gene on 5q35 to a novel anaplastic lymphoma kinase (ALK) on 2p23.6 As a consequence of this translocation, the NPM and ALK genes are fused to form a chimeric transcript that encodes a hybrid protein (p80) in which the aminoterminus of NPM is linked to the catalytic domain of ALK. Two distinct oncogenic effects are thought to be caused by the t(2;5) translocation. First, the ALK gene, which is not physiologically expressed in normal T lymphocytes, undergoes heterologous expression in lymphoma cells, conceivably because of its juxtaposition to the promoter sequences of NPM, which are physiologically expressed in T cells. Second, based on the activation model of other tyrosine kinase oncogenes, one would predict that the truncated ALK constitutively phosphorylates intracellular targets to trigger malignant transformation. The pathogenetic role of NPM/ALK rearrangements is supported by studies in vitro and in vivo. In particular, retroviral-mediated gene transfer of NPM/ALK in vivo causes T-cell lymphoid malignancies in mice.48
III. DNA Microarray Analysis: What Can It Tell Us about the Biology of Lymphoid Neoplasms?
Wing C. Chan, MD*
Department of Pathology, University of Nebraska Medical Center, Omaha NE 68124
Rationale for gene expression profiling of cancers
Carcinogenesis is generally initiated by a genetic lesion that results from an error occurring during normal cell function or from unrepaired physical or chemical damage to the genome.1 Rarely, the abnormal gene is inherited, resulting in an increased susceptibility to cancer for all family members who have inherited the gene.2 This initial event provides an increased chance for additional genetic lesions to develop, usually over a number of years. When a cell acquires the proper combination of genetic lesions, it will have the full potential to generate a malignant tumor. As the neoplastic cells continue to divide and expand, additional genetic alterations may be acquired and some of these may contribute to characteristics that make the tumor more clinically aggressive and/or resistant to treatment, such as enhanced growth rate, independence of growth signals and resistance to death-inducing signals.
We can postulate that the characteristics of a tumor and its clinical behavior are determined by the unique set of genetic lesions harbored by the tumor cells. These genetic lesions alter the pattern of mRNA expression in the cell, and this altered pattern can be regarded as the “molecular signature” or “fingerprint” of the tumor. Tumors with closely related genetic lesions will have very similar “signatures” and also will be expected to have similar clinical behaviors. It is, therefore, logical to make the following assumptions: 1) gene expression profiling will help us establish a clinically relevant and biologically meaningful cancer classification; 2) gene expression profiles will be helpful in prognostication and treatment decision making in individual cases; and 3) studying gene expression profiles will make it possible to identify genes that are important determinants of the behavior of lymphomas.
Techniques for Gene Expression Profiling
One way of obtaining the gene expression profile of a tumor is to prepare a cDNA library from the extracted mRNA and perform a massive sequencing of the clones. This approach is useful for gene discovery but not practical for profiling a large series of tumors. A more efficient method has been developed: Serial Analysis of Gene Expression (SAGE), which still involves a complicated series of experimental manipulation and a substantial amount of DNA sequencing for each specimen.3,4
The DNA microarray is, in principle, a reverse Northern blot, in which the “probes” for various mRNA species are immobilized on a solid support, and the sample to be examined is labeled and hybridized to the immobilized probes. The intensity of the hybridization signal on each probe is related to the concentration of the corresponding mRNA in the sample. A DNA-microarray usually contains thousands of immobilized probes, most commonly on a solid non-porous support, although membrane based arrays are still used by some investigators. The recently revised estimate of the number of human genes is somewhere around 30,000,5,26 but there are more mRNA species that arise through alternative splicing and other mechanisms. The current collection of over 40,000 human cDNA clones at Research Genetics (http://www.resgenm.com) constitutes a substantial proportion of all mRNA transcripts. While most investigators are not using all of these clones in their experiments, an array with 10,000 clones, especially if they are enriched with known genes and expression sequence tags (ESTs) of interest for a particular investigation, will be able to monitor the expression of a major population of genes.
Two platforms of DNA microarrays are commonly used. In one, the spots on the microarray consist of polymerase chain reaction (PCR) amplified products of the cDNA inserts in plasmid clones. After appropriate preparation, these PCR products are spotted on a poly L-lysine or aminosilane coated glass slide using an arrayer.7,8 The other type of microarray contains oligonucleotide probes. In the one manufactured by Affymetrix Inc., the oligonucleotides are synthesized in situ by a process called photolithography.9 It is also possible to synthesize oligonucleotide probes off chip and then attach them on microarrays; new methods of in situ synthesis have also been described.10 The advantage of the cDNA microarray is the flexibility of design, which allows more ready customization of the array to fit the needs of the investigators. If a large number of arrays are needed, it may be more economical to produce them in an array facility than to purchase commercial arrays.11 On the other hand, the fabrication of the cDNA arrays and the quality controls are quite variable among laboratories. Even for cDNA microarrays produced in the same facility, the spots on the array are not completely uniform and may vary significantly within and between arrays. Therefore, hybridization is typically performed with the addition of a standard RNA preparation to the test RNA. The standard and test RNA samples are labeled by reverse transcription with different fluorescent dyes. The ratio of the test versus standard cDNA hybridized to each of the spots depends on their relative concentration in the mixture. The fluorescence due to the test and standard cDNA on each spot is quantitated and expressed as a ratio. The concentration of each cDNA in different test samples can, therefore, be compared with each other because the measurements are all expressed as a ratio of the standard. There have been extensive discussions on the merit of using a universal standard so that results can be readily compared across different laboratories, both for scientific exchange and for quality assessment of the microarray experiments.
Data Management and Analysis
For each microarray experiment, there are thousands of experimental measurements that need to be processed, including fluorescence measurement, background subtraction and data normalization.8 It is desirable to perform each experiment in duplicates or triplicates in order to differentiate between experimental variations and real differences in expression level. However, it is often not possible to obtain sufficient amounts of RNA in clinical settings for multiple experiments, and the cost involved is also a consideration.
After image processing, a massive amount of information must be analyzed. A number of analytical tools are currently available for detecting structures in the data set, for model fitting, class prediction/assignment and class discovery.12–,17 There is no single best tool, and the most appropriate tools for an experiment depend on the experimental design, the data obtained and the questions being addressed. A detailed discussion of the analytical methods is beyond the scope of this communication and only a brief outline of one of the most widely used tools—agglomerative hierarchical clustering—will be presented.13 The most common format for presenting gene expression analysis is in the form of a matrix, with a list of genes on the microarray (usually on the Y-axis) plotted against the tumor samples (usually on the X-axis) in the study. Agglomerative hierarchical clustering is a “bottom-up” clustering method, starting by clustering pairs of genes with the most similar pattern of expression across samples, and successively combining these initial clusters into larger clusters until all the genes are clustered into a dendrogram (Figure 8 [color page 545] and Figure 9 ). Samples can be similarly clustered according to their overall similarity in gene expression profiles. Clustering can be performed either unsupervised or supervised. In unsupervised clustering, the predefined clustering algorithm is allowed to arrange the genes and samples. In supervised clustering, certain investigation-defined parameters, based on some prior knowledge, are employed to guide the clustering. These parameters may be, for example, clinical data or a set of genes with certain known biologic functions.
Gene expression profiling is a powerful technology, but additional information on the tumor, including cytogenetic/molecular genetic data, and on the patients can markedly enhance the discovery process. Depending on the questions to be addressed, a sufficient number of cases should be included to allow statistically meaningful analysis. Validation of the analytical results is important. Different analytical tools may be applied to confirm the validity of the conclusions from one analytical method. The reproducibility of clustering may be tested by introducing random Gaussian noise to each data point, and the perturbed data are then re-clustered. A validation set of samples may be analyzed to test the conclusion drawn from a prior experiment with a different set of samples.
Special Considerations in the Study of Human tumor Specimens
A number of issues confront investigators studying clinical specimens. Typically, the specimens have not been collected in a uniform, controlled fashion, thus introducing certain variables that may influence the gene expression pattern. In many of the studies, the tumor samples are stored as frozen tissues, so that separation of tumor from other non-neoplastic elements at the time of the study is not practical. When whole tissue is used for microarray analysis, the gene expression profile is a composite of all types of cells present. This complicates the interpretation of the data but, on the other hand, there is added information on the gene expression profile of the infiltrating lymphocytes, macrophages and stromal cells. The pattern of host response may provide important insight into the biology of the tumor and the clinical course of the patient. To assess the expression level of selected genes in tumor cells, specific analysis such as RT-PCR on micro-dissected tumor, in situ hybridization or immunohistochemistry may be performed. It may also be possible to obtain sufficient tumor cells by micro-dissection of frozen tumor sections for total cDNA amplification for confirmatory microarray analysis.18,19
Gene Expression profiling in Human Malignancies: Current Status
There are numerous ongoing gene expression profiling studies on many different types of human malignancies. One of the major themes in these experiments is to explore the potential of gene expression analysis in class prediction (classifying tumors into currently defined categories) and class discovery (finding new tumor types that are biologically meaningful). There are good indications that gene profiling will be successful in both. The promises and challenges gleaned from these early studies are discussed below using illustrative examples.
Alizadeh et al20 studied three types of B-cell malignancies: DLBCL, follicular lymphoma (FL) and B-chronic lymphocytic leukemia (CLL), using a microarray enriched in genes known to be involved in lymphoid neoplasms and in lymphocyte biology. Using unsupervised hierarchical clustering, these three categories were separated broadly into three corresponding clusters according to their overall gene expression pattern. This suggests that distinct groups of lymphoma defined by traditional parameters have sufficiently different patterns of gene expression that they can be separated by the set of genes examined on the array (class prediction).
A major potential pitfall in this interpretation was that different types of specimens were used for the different diseases. The DLBCL were submitted as frozen tumor tissue, while many of the FL and CLL samples were comprised of enriched tumor cells; therefore, there was differential expression of large sets of genes associated with stromal elements and infiltrating T-cells in the tumors. In addition, DLBCL generally has a higher proliferation rate than FL and CLL and, hence, exhibits up-regulation of genes associated with cell proliferation. The common expression of these large sets of genes may move cases into the same cluster despite the presence of important biologic differences. Therefore, additional analyses taking into consideration all important confounding variables are necessary.
The investigators noticed that there was a set of genes preferentially expressed by normal germinal center (GC) B-cells but not by peripheral blood B-cells activated by a number of stimuli. When the set of GC-B cell associated genes was used to cluster the DLBCL cases, two broad groups were delineated. One group expressed many of the genes in the GC-B-cell associated profile, while the other expressed few of the GC-B-cell associated genes but, instead, expressed many of the genes on the activated B-cell profile. Hence, two subgroups of DLBCL that appear to be biologically distinctive can be defined by their gene expression profile (class discovery).
Golub and colleagues21 studied acute myeloid (AML) and acute lymphoid leukemias (ALL) using an Affymetrix array and found that class prediction of most cases of AML, T-ALL and B-ALL could be accomplished. Interestingly, many of the genes useful in class prediction of AML versus ALL are not lineage-specific markers, indicating that differences in the biology of the tumor cells that go beyond lineage differentiation. A workshop was held at Duke University (Dec, 2000) where the set of data generated by Golub et al was analyzed by the participants, and most groups were able to accurately predict all but one of the test cases based on the expression data.22
Similar class prediction and/or class discovery studies have been performed on other tumors, including malignant melanoma,17 breast carcinoma23,37 and childhood sarcoma.24
When apparently new tumor categories are discovered on analyzing gene expression data, the finding requires careful validation. Aside from reanalyzing the data using various tools, one can examine these new classes for biologic and clinical relevance using independent parameters other than gene expression. Alizadehs et al20 examined the clinical distinctiveness of the two new classes of DLBCL by correlating the overall survival (OAS) of the patients with the microarray classification. A significantly better OAS was associated with the group of lymphoma with the GC-B cell-like profile and this association appeared to hold even when cases with low clinical risk factors (IPI of < 3) were examined. The clinical data thus provided independent support for the validity of the class discovery. A recent study by Shipp et al25 directly examined the usefulness of gene expression profiling using the Affymetrix array in predicting clinical outcome in a series of patient with DLBCL who had received CHOP-based chemotherapy. Two groups with significant differences in survival could be identified within the entire population and also within patients in the intermediate IPI risk categories. It would be highly interesting to confirm the validity of these findings with an independent set of cases.
The validity of the class discovery can also be queried by independent biologic parameters. One cardinal feature of GC-B cells is the presence of ongoing somatic hypermutation of the immunoglobulin (Ig) genes. The group of DLBCL with an expression profile similar to GC-B cells would be expected to exhibit this characteristic, while the other group should not. This hypothesis was tested in 14 of the cases previously studied by Alizadeh and colleagues.20 All 7 cases with the GC-B-like gene expression profile showed ongoing somatic hypermutation of their IgH genes, while only 2 of 7 of the cases with the activated B-cell-like pattern showed ongoing mutations, but at a lower level compared with the previous group.26 The 2 cases with unexpected ongoing mutations were at the junction of the two large clusters and may represent cases with overlapping biologic characteristics, which could explain the unexpected behavior.
Since the unique gene expression profile of a tumor is determined by the intricate interaction of the genetic abnormalities present, it is anticipated that each of the genetic abnormalities, especially those with a major influence on the biology of the tumor, will leave some unique “footprint” on this profile. Gene expression signatures correlating with specific genetic abnormalities may be detectable. The t(14;18)(q32;q21) is a hallmark of FL but is also detectable in 20-30% of cases of de novo DLBCL. If the bc1-2 translocation is an initiating event for DLBCL as for FL, one would expect that the precursor cells of these large cell lymphomas would also start their journey in germinal centers. Different secondary events lead to alternative pathways resulting in either FL or DLBCL. It is likely, therefore, that DLBCL with t(14;18) would exhibit the GC-B cell-like expression profile. A recent study demonstrated that this is indeed the case.27 Seven of 35 cases of DLBCL had t(14;18) and all of these had the GC-B cell gene expression profile. Six of 7 cases also clustered closely with each other, with normal GC-B cells and a cell line with t(14;18). This finding supports the validity of the sub-division of the DLBCL into two major subtypes according to their gene expression profile, and it also supports the contention that significant genetic alterations may be associated with identifiable expression signatures.
In a recent report,28 the gene expression profiles of sporadic breast carcinomas and cases with mutation of BRCA-1 or BRCA-2 were studied to determine whether tumors with BRCA-1 or -2 mutations were identifiable through differences in the expression profiles. Groups of genes with significant differential expression in tumors with the two different mutations were identified, supporting the hypothesis that important genetic alterations may be associated with unique gene expression profiles.
The above findings underscore the importance of determining the genetic abnormalities in tumors submitted for gene expression profiling. Genetic abnormalities are frequently not readily apparent from the expression profile and unlikely predictable from the expression level of the corresponding mRNA alone. A careful correlative study between known genetic abnormalities and the corresponding gene expression profiles is essential to discover the expression footprints associated with specific genetic lesions. Routine cytogenetic data may be significantly enhanced by multicolor karyotyping such as SKY and M-FISH.29,30 Additional information may be obtained by comparative genomic hybridization30– 32 or FISH analysis of specific loci. Abnormalities in specific genes such as mutations and methylation may also be determined by a number of molecular assays. This information will be very helpful in the interpretation of gene expression data, which, in turn, may help us understand the functional effect of a genetic abnormality.
At this stage, few genes or pathways have been identified by gene profiling studies of tumors to be essential components in defining clinical behavior and/or various biologic characteristics. In general, there are hundreds or more genes that are differentially expressed at various levels between even closely related categories of tumors. To identify the important versus the secondary or accompanying events is a major challenge. Gene expression data should not be interpreted in isolation. All ancillary information including various tumor and clinical characteristics could be helpful in their interpretation. A few candidate genes have been proposed. The RhoC gene has been implicated to be a key determinant of metastatic potential and tumor invasion in melanoma cells.33 Down regulation of c-MYC and IL-6 expression has been shown in myeloma cells exposed to thalidomide, and these are thus believed to be important target genes for the drug.34 There will be an exponential increase in candidate genes in the next few years, and the painstaking task of confirming their importance and delineating the mechanisms of action will have to follow.
Numerous attempts have been made to develop assays that will accurately predict the sensitivity of a tumor to various chemotherapeutic agents. The possibility that the gene expression pattern of a tumor may provide the chemo-responsiveness profile has been examined in some preliminary experiments using a collection of cell lines (NCI-60). The results are promising, and suggest that selecting the most appropriate therapy based on gene expression profile is feasible.35,36
Perspective
Gene expression profiling of cancer has shown tremendous promise in delineating the molecular mechanisms and the key genetic components underlying the different biologic properties and clinical behaviors among malignant neoplasms. Some of these genes and their products may be suitable targets for therapeutic intervention. The treatment of many types of cancers is limited by the lack of new, effective drugs. It is hoped that novel agents will be developed based on the molecular targets identified. While it is useful to stratify patients to the most appropriate therapeutic regimens based on their individual risk factors, the choice of therapy and the understanding of the biologic basis underlying these risk factors are currently limited. When novel, mechanism-based therapies become available, it will be essential to have the relevant molecular information for each tumor. One can envision the development of diagnostic microarrays containing all the essential genes, which have been selected based on knowledge gained from prior gene profiling studies. Every tumor could be examined by the relevant microarray at diagnosis, and the results would help to determine the appropriate therapeutic interventions. Comprehensive molecular tumor diagnostics and individualized treatment may become a reality in the not-too-distant future.
IV. Why Do I Need to Know This? Implications of the New Biology of Lymphomas for Clinical Practice
Nancy Lee Harris, MD*
Department of Pathology, Warren 2, Massachusetts General Hospital, 55 Fruit Street, Warren 219, Boston MA 02114
Physicians who treat lymphoma patients are fond of recalling the “good old days,” when there were only four types of lymphoma: lymphosarcoma, reticulum cell sarcoma, follicular lymphoma, and Hodgkin's disease. Many of my oncologist colleagues complain bitterly about the seemingly endless expansion of lymphoma subtypes and the mind-numbing complexity of nomenclature in recent years. These same individuals, however, seem to have no problem digesting and absorbing what seems to a pathologist like a bubbling cauldron containing an ever-changing alphabet soup of drug names and doses used to treat these same diseases. Why are two groups of supposedly intelligent scientists so incapable of understanding each other's language? The reason has nothing to do with intelligence: it's simply a matter of “need to know.” Pathologists need to sort and classify specimens according to some criteria that permit them to organize and learn a vast array of tumors. Oncologists need to understand the treatments they deliver to patients in their care. Neither feels this sense of personal need when it comes to the other's language.
When pathologists and clinicians speak the same language, real advances can occur. Two examples illustrate the power of translation between the language of physicians who diagnose and classify malignancies and the language of those who treat them. In the 1980s, a new category of lymphoma—derived from mucosa-associated lymphoid tissue (MALT lymphoma)—was discovered by careful morphologic and immunophenotypic analysis.1 This type of lymphoma, unlike any of the other known “low-grade” lymphomas with which it had been lumped, was shown by a pathologist in the 1990s to be related to an immune response to a bacterium—Helicobacter pylori.2 This led to the discovery that many cases can apparently be cured simply by eradicating the infection with antibiotics.3,4 Thus, a pathologist took a disease full circle, from discovery to cure. In 1994, when the REAL classification was published (Table 5 ), it was criticized as a pathologists' game—a bunch of new disease categories without proven clinical relevance.5,6 One oncologist who understood the language of pathology organized a worldwide clinical test of the classification, showing that it defined new diseases with distinctive clinical features and responses to treatments, thereby convincing oncologists to accept it.7 Thus, an oncologist took a pathologic classification from theory to clinical practice. In the past two years, the gap between basic science and clinical practice has been dramatically bridged by the discovery that the abnormal protein produced by the BCR/ABL translocation of chronic myelogenous leukemia could be inhibited by a specifically designed drug, resulting in remissions in many patients with this disease.8 This principle could be extended to some categories of lymphoma characterized by fusion proteins that activate tyrosine kinases, such as anaplastic large-cell lymphoma. These improvements in clinical practice could not occur without the combined efforts of pathologists and oncologists to define distinct categories of disease whose pathophysiology and genetics can then be studied as targets for therapy.
One reason that hematologists who treat leukemias are interested in different disease subtypes and in classification by genetics is that they can see in their own patients how the clinical behavior of the disease is predicted by these features and the advantages of tailoring their treatment to specific disease categories. For physicians who treat lymphomas, the clinical differences between the most common diseases are easily appreciated—large B-cell lymphoma, follicular lymphoma, chronic lymphocytic leukemia (CLL), and Hodgkin's disease. Many of the more recently recognized diseases are also clinically distinctive—MALT lymphoma and mantle cell lymphoma are dramatically different from each other and from CLL/small lymphocytic lymphoma, and require totally different approaches to treatment. Peripheral T-cell lymphomas comprise many distinctive categories that differ from aggressive B-cell lymphomas. As practitioners begin to see patients with these diseases, their distinctive clinical features will provide a real-life rationale for learning these new disease categories.
But most of these newer diseases are relatively uncommon and comprise a minority of any oncologist's practice. Thus, recent studies of immunophenotype and genetics have resulted in defining new disease categories that are distinctive but rare, and—paradoxically—not in defining clinically distinctive subsets or new approaches to therapy for the common diseases such as large B-cell lymphoma and follicular lymphoma. Interestingly, however, one common disease—chronic lymphocytic leukemia—has recently been found to comprise two genetically distinct subtypes: those without immunoglobulin variable region gene mutations (naïve or pre-germinal center B cells) and those with mutated IgV region genes (post-germinal center or memory B cells).29 These two types of CLL have different clinical behavior, with the cases corresponding to naïve B cells having much shorter survival than the memory B cell cases. Further dissection of the more common diseases into prognostically distinct groups, using new techniques such as microarray analysis of gene expression, will be a focus of the next decade.
In the meantime, understanding the relationship of lymphoid neoplasms to their normal counterparts, the genetic events that lead to malignant transformation in lymphoid cells, and the novel technology of DNA microarray analysis are essential for practitioners involved in the care of patients with these diseases.
B-cell Differentiation: Why do I need to know this?
The vast majority of lymphoid neoplasms seen by practitioners in the United States and Western Europe derive from B cells at various stages of differentiation and possess many features of their normal counterpart (Table 6 ). These features—including normal genetic events, gene expression, immunophenotype, morphology, homing patterns, and proliferation fraction—in large part dictate the clinical behavior of these diseases. These biological features of normal B cells can help us understand many things about lymphomas (Table 7 , Table 8 ; Figure 10, color page 546).
Clinical features of lymphomas
Who will get the disease? Typically patients with large pools of the normal cell type, in which neoplastic transformation can occur: for example, lymphoblastic neoplasms are more common in children who have large pools of precursor B cells; plasma cell myeloma is common in older adults with large pools of post-germinal center antigen-exposed plasma cells; and marginal zone lymphomas are common in patients with autoimmune diseases and intestinal infections.
How will the tumor behave?Tumors corresponding to proliferating normal cells such as lymphoblasts and centroblasts tend to be rapidly growing and “aggressive,” while those corresponding to resting stages, such as CLL/SLL, tend to be indolent.
Where will the tumor grow?Tumors of marrow-derived precursor cells become acute leukemia and those of marrow-homing plasma cells multiple myeloma; tumors of germinal center cells populate lymphoid follicles throughout the body, and tumors of MALT pop up in non-contiguous extranodal sites.
How does the disease develop?The genetic events that occur in B-cell differentiation, involving rearrangement and mutations of the immunoglobulin genes, provide the substrate for most of the genetic accidents that result in the development of B-cell neoplasms—translocations or mutations involving the immunoglobulin gene loci.
Classification and diagnosis
Pathologists exploit the biological features of normal and neoplastic B cells for diagnosis: a combination of morphology, normal gene expression as recognized by immunophenotype, genetic alterations such as receptor gene rearrangement and mutation, and clinical features are used to define the lineage and differentiation stage of the tumor. Antigens differentially expressed at stages of lymphocyte differentiation and activation are essential for modern diagnosis and classification: these include pan-B and pan-T antigens, precursor-associated antigens such as TdT, antigens associated with naïve B cells, such as CD5, the germinal center, such as CD10 and bcl6, or post-germinal center cells, such as CD38 and CD138. Detection of immunoglobulin and T-cell receptor gene rearrangement by molecular genetic techniques such as Southern blot (which requires fresh tissue) and PCR (which can be done on paraffin-embedded tissue) can be a useful adjunct to morphology in distinguishing between atypical reactive processes and lymphomas, and for classifying difficult cases as T or B lineage neoplasms. (Since antigen-receptor gene rearrangement is not always lineage specific nor an indication of neoplasia, these results must be interpreted with caution.) The homing patterns of the normal cells are also exploited for diagnosis: an extranodal lymphoma with plasmacytic differentiation is likely to be a MALT lymphoma, while a morphologically similar infiltrate in the bone marrow would be more likely to be a lymphoplasmacytic lymphoma, associated with Waldenström's macroglobulinemia.
Understanding and developing treatments
Oncologists exploit the biological features of lymphoid neoplasms to devise therapies—for example, tumors of rapidly proliferating cells typically respond to the current armamentarium of drugs that interrupt DNA synthesis. Potentially, as with the MALT lymphoma story, a better understanding of the specific features that drive proliferation and survival of some of the other low-grade lymphomas may lead to additional novel therapies. For example, adherence to follicular dendritic cells and interaction with T cells through surface ligands and chemokines may be important for the survival of follicular lymphomas, as it is for normal germinal center cells —disrupting these interactions may have potential therapeutic utility. Lymphoblasts, CLL cells, and myeloma cells all depend on interaction with marrow stromal cells, and investigation of these interactions may lead to new therapeutic options for these diseases.
Molecular Genetics of Lymphomas: Why Do I Need to Know This?
Most lymphoid neoplasms have genetic abnormalities. The discovery of specific translocations has been important in defining certain diseases, such as Burkitt's lymphoma, follicular lymphoma, mantle cell lymphoma, and anaplastic large-cell lymphoma. Most others occur only in a subset of the cases of the disease or are not entirely specific for the disease—even the t(8;14) of Burkitt's lymphoma and the t(14;18) of follicular lymphoma may be found in large B-cell lymphoma, and the t(11;14) of mantle cell lymphoma occurs in plasma cell myeloma. These genetic abnormalities can help us to understand the pathogenesis of lymphomas, they can help in disease definition (classification) and diagnosis, and they can be exploited for treatment.
Understanding pathogenesis
Chromosomal translocations have provided important information about the pathogenesis of lymphomas; analysis of the genes involved in the breakpoints has shown that most involve genes associated with either proliferation or apoptosis. The vast majority of the translocations in lymphoid neoplasms involve placing a proto-oncogene under the control of a promotor associated with an antigen receptor gene—either one of the immunoglobulin genes or one of the T-cell receptor genes. A few lymphoid neoplasms have translocations that produce fusion proteins, which are more characteristic of the myeloid leukemias. These translocations are thought to occur predominantly in precursor cells, as accidents in the normal rearrangement of antigen receptor genes. However, particularly in B cells, some of the translocations may occur at the germinal center stage, when the somatic mutation that occurs during affinity maturation introduces DNA strand breaks. Both the stage at which the translocation occurs and the specific translocation are probably important in determining the nature of the subsequent malignancy.
Classification and diagnosis
The discovery of specific translocations has helped to define distinct disease entities to add to classifications of lymphoid neoplasms—what the microarray people are now calling “class discovery.” For example, investigation of the t(11;14) showed that it was consistently associated with a small B-cell neoplasm that had been categorized differently in different classifications or not recognized at all; this disease came to be known as mantle cell lymphoma and is now considered an important and clinically distinctive category of B-cell lymphoma.10,11 Similarly, the discovery of the t(2;5) in a subset of what was initially called “malignant histiocytosis” led to the recognition that it defined a clinically distinctive category of T-cell lymphoma, now known as anaplastic large-cell lymphoma.12,13 When a lymphoma is associated with a specific translocation, detection of the translocation can be useful in diagnosis of a specific patient —both for distinguishing lymphoma from a benign lymphoid process and in making a specific lymphoma diagnosis—what the microarray people call “class prediction.”
Translocations can be detected by genetic techniques —classical cytogenetics, Southern blot, or, increasingly, PCR analysis of paraffin-embedded tissue—but also by immunohistochemistry in many cases. An important consequence of many translocations is overexpression of a protein not ordinarily found in that cell type. Thus, rather than requiring genetic analysis, the genetic abnormality can be detected by immunohistochemistry. For example, in follicular lymphoma, the differential diagnosis often includes reactive lymphoid hyperplasia. Normal germinal center cells down-regulate bcl2 protein to make them susceptible to apoptosis in negative selection. Lymphomas with the t(14;18) and BCL2 rearrangement express this protein; thus, detection of bcl2 in a follicle is evidence that it is neoplastic.14 Bcl2 expression in follicles can also be used to distinguish follicular lymphoma from MALT lymphoma with follicular colonization, in which normal bcl2-negative follicle center cells are typically present. However, since normal resting (non-germinal center) B cells and most small B-cell neoplasms express bcl2, its expression on the neoplastic cells is not useful in classification of small B-cell neoplasms. In mantle cell lymphoma, the t(11;14), results in overexpression of cyclin D1, a cell-cycle protein not normally expressed in lymphoid cells. This protein can also be detected by immunohistochemistry and is useful in distinguishing mantle cell lymphoma from diffuse reactive lymphoid infiltrates and, more importantly, from other types of small B-cell lymphomas.15,16 The other major translocation associated with diagnostically useful gene expression is the t(2;5) of anaplastic large-cell lymphoma, which results in expression of the ALK protein, not normally expressed in lymphoid cells. This protein can also be detected by immunohistochemistry and is essential for the diagnosis of ALCL.17,18 Finally, MALT lymphomas with the t(1;14) can be recognized by their overexpression of bcl10 protein, which can be detected by immunohistochemistry; these cases may have a worse prognosis. In addition, cases with the t(11;18), about 40% of gastric MALT lymphomas, also overexpress bcl10 protein; this translocation is associated with failure to respond to antibiotic therapy19– 21 (Table 9 ).
Detection of genetic abnormalities by genetic techniques remains an essential diagnostic tool in many situations. In this era of gene expression profiling, it is important to remember that detection of specific genetic abnormalities may provide important diagnostic and clinical information above and beyond that provided by analysis of mRNA or protein. Many translocations do not result in abnormal protein expression that is diagnostically useful. The c-MYC/Ig translocation of Burkitt lymphoma leads to MYC overexpression, but the protein is expressed in many normal cells and in lymphomas lacking the translocation, so is not diagnostically useful. Similarly, BCL6 and PAX-5 translocations lead to overexpression of the corresponding protein, but many B cells without the translocation normally express these proteins. Other abnormalities such as trisomies and deletions have not yet been associated with specific genes or proteins that can be exploited for diagnosis. In addition, genetic techniques to detect translocations associated with specific protein expression can be useful in diagnosis in some cases. For example, since normal resting B cells and neoplasms derived from them typically express bcl2, expression of bcl2 protein on neoplastic cells is not useful in classification – virtually all small B-cell lymphomas will express it, whether or not they have a t(14;18).22 Detection of cyclin D1 by immunohistochemistry is not always reliable and cannot be used on some types of specimens. In all of these situations, detection of the genetic abnormality by classical cytogenetics, FISH, Southern blot or PCR on paraffin-embedded tissue is useful both in confirming the neoplastic nature of a lymphoid proliferation and in subclassification of a lymphoid neoplasm.
Staging and prognosis
Genetic abnormalities can also be exploited in staging and in detection of minimal residual disease; for example, PCR for the t(14;18)/BCL2 rearrangement is used in follow-up of patients undergoing bone marrow transplant for follicular lymphoma, and immunostaining or FISH for cyclin D1/BCL1 rearrangement can be used to follow patients with mantle cell lymphoma. Tumor-specific probes for rearranged immunoglobulin genes have also been used to detect minimal residual disease in B-cell lymphoma patients.
Genetic abnormalities may also affect prognosis and can be used to identify patients with differing expectations of outcome. These include trisomy 12 and abnormalities of 13q in CLL, which confer a worse and better prognosis respectively, t(11;18) in gastric MALT lymphoma, which is associated with a decreased likelihood of response to antibiotic therapy and with a decreased frequency of transformation to large-cell lymphoma,19 and the t(2;5) in ALCL, which is associated with a good prognosis.
Developing new therapies
Finally, the future hope is that some of these genetic abnormalities can be exploited for the development of new therapies, analogous to the STI571 story in CML. Antisense oligonucleotides to anti-apoptosis genes such as BCL2, drugs that target fusion proteins such as NPM/ALK or API2/MLT1, or drugs that inhibit cell cycle proteins such as cyclin D1 are all possible areas to explore for new treatments of these diseases.
DNA Microarray Analysis: Why Do I Need to Know This?
DNA microarray analysis is essentially a way of fishing for information that would take much longer if we looked for one gene at a time. The information is only as good as the selection of DNA to place on the array and the choice of software to analyze the resulting data.23,24 Like most new techniques—cytogenetics in the 1960s, immunophenotyping in the 1970s and 1980s and molecular genetic analysis in the 1980s and 1990s—it is exciting and seemingly unlimited in its potential to revolutionize the diagnosis and classification of tumors and define prognosis for individual patients. But as with all technical breakthroughs that preceded it, we have to do our homework with this technology to really find out what it can do for us. Practicing oncologists need to understand this technology for the following reasons: 1. to have a sense of its potential, 2. to know its limitations, and 3. to know how it fits with other diagnostic and prognostic tools. I will focus here on its practical limitations and how it can dovetail with other techniques.
Classification and diagnosis
Proponents of DNA microarray technology now look forward to a purely genetic classification of lymphoid neoplasms, much in the way that we optimistically spoke of a classification based purely on monoclonal antibodies in the 1980s. Previous experience would suggest that diseases will continue to be defined by a combination of parameters, including genetic features (both normal changes related to lineage and differentiation and abnormal changes related to translocations and other alterations), changes in gene expression as detected by protein expression (immunophenotyping), and clinical features. Microarray analysis is simply a technical method for studying the expression of many genes at once at the messenger RNA level.
For disease definition (class discovery), one would theoretically put as many genes as possible on a chip and compare expression among unknown samples of tumors, using statistical analysis to identify tumors with similar expression patterns followed by clinical observation to see whether the categories thus defined were distinctive. Obviously, this approach contains many moving targets, including the choice of genes to study, the purity of the original specimens, the treatments given to the patients, and the endpoint for deciding what clinical differences are important. For this reason, most microarray studies to date have focused more on diagnosis (class prediction) and prognosis (a more restricted form of class discovery) in a defined subgroup of cases. As reported by Dr. Chan, several studies of lymphomas and leukemias have shown that microarray analysis can classify most cases of leukemia and lymphoma into previously defined categories based on expression of genes known to be associated with lineage and differentiation stage,25 and that within one major categorylarge B-cell lymphoma—prognostic groups can be defined.26
How will this be used in practice?
If combinations of genes associated with particular diseases or prognostic groups can be identified and placed on a chip, there is the potential for rapid screening of patient specimens for diagnosis and prognosis. In reality, the ability to do this effectively in routine practice is probably at least 5 and possibly more years in the future. A more immediate potential is for studies already underway to reveal novel genes associated with prognostically important categories that can be assessed in individual specimens by traditional methods. The product of an expressed gene is a protein, and overexpression of normal proteins or expression of abnormal proteins can be readily assessed by immunofluorescence or immunohistochemistry. Many of the genes associated with the germinal center and non-germinal center categories of Alizadeh and associates were already known and can be studied by immunohistochemistry—these include CD10, BCL6 and BCL2.26 Other genes associated with these two prognostically important groups have not been studied and should be the focus of future research.
Even more traditionally, we should remember that morphology is an important manifestation of gene expression. Traditional morphology defined the same two prognostically distinct categories of large B-cell lymphoma in 1974 that were rediscovered by microarray analysis in 2000! Pathologists' eyes can be trained and our vision refined by looking at slides of cases that have been shown by novel techniques to have a particular characteristic. The recognition of anaplastic large-cell lymphoma, for example, was stimulated by staining for CD30 (Ki-1) and the discovery that cases that showed strong staining had a common and characteristic morphology.27 The recognition of lymphocyte-rich classical Hodgkin's lymphoma followed immunostaining of a large group of cases previously diagnosed as lymphocyte predominant type for antigens associated with classical Hodgkin's lymphoma (CD15 and CD30) – this category can now be recognized by morphology by the same pathologists who were unable to “see” it before the study.28,29 Pathologists have long debated the feasibility and clinical relevance of morphologic subclassification of large B-cell lymphoma into germinal center (centroblastic) and non-germinal center (immunoblastic) types, as prescribed by the Kiel Classification.30 Possibly morphologic review of cases shown by gene expression to fall into one or the other category will reveal previously unappreciated morphologic features that will permit them to be reproducibly diagnosed by morphology alone.
It is entirely possible—and maybe desirable—that in the future all lymphomas will be diagnosed simply by doing a fine needle aspirate, extracting RNA or protein and subjecting the product to high throughput genomic or proteomic analysis, which will distinguish benign from malignant infiltrates, lymphomas from other neoplasms, and stratify lymphomas and other tumors into specific categories for therapy. Until then, using the information generated by research using these techniques to improve traditional diagnostic methods may yield immediate benefit for patients (Table 10 ).
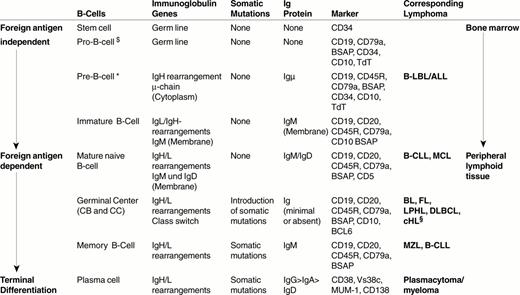
Events in the rearrangement of immunoglobulin genes.
| Rearrangement of the Immunoglobulin heavy chain (IgH) gene |
| • The first step occurs in the “early pre-B-cells” and involves the recombination of the D-(Diversity) and J-(Joining) segments of the heavy chain genes (IgH). During this process, one of 27 possible D-segments and one of 6 possible J-segments are randomly chosen and fused closely together, resulting in exclusion of the DNA segments which previously separated them (see Figure 2). In addition, nucleotides of variable length and composition are randomly inserted between the rearranged D- and J-segments (N′-region). |
| • The second step occurs in the “late pre-B-cells” and involves the rearrangement of the V-(Variable) segment, resulting in the complete rearrangement of the IgH gene (Figure 2). It involves the insertion of one of a possible 59 V-segments with the above-formed D-J-segment and the exclusion of the DNA segments which previously separated them (see Figure 2). In addition, as above, nucleotides of variable length and composition are randomly inserted between the rearranged D-J- segments and the V-segment (N-region). In this way, a different DNA segment (V-N-D-N′-J) for each B-cell is created, which serves as a “finger-print” for it and all of its' daughter cells. |
| Rearrangement of the Immunoglobulin light chain (IgL) gene |
| •The rearrangement of the two IgL-genes (κ and λ) also takes place in the pre-B-cell, and occurs in a similar manner to the rearrangement of IgH. In contrast to the IgH, the IgL-genes do not possess any D-segments. As a consequence the rearrangement of the IgL involves only one step, whereby nucleotides of variable length and composition (N-sequence) are inserted between the V- and J-segments. In this way, a second DNA segment (V-N-J) is created, which differs between each B-cell. When the Ig-gene rearrangements are completed the immature B cells have evolved. |
| Non-functional Ig-rearrangements |
| • Through the above-mentioned randomised insertion of nucleotides during the IgH and IgL rearrangements, “Stop-codons” or “Frame-shifts,” which inhibit Ig-protein functioning, occur relatively frequently. In such cases, a second independent rearrangement of either the IgH or IgL occurs. Should this additional rearrangement result in a non-coding DNA sequence and, therefore, to a non-functional Ig receptor molecule, the B-cell is eliminated via apoptosis. |
| Rearrangement of the Ig genes in normal and lymphomatous B-cells |
| • As described above, every cell which arises from progenitor B-cells possesses two Ig gene rearrangement products, VH-N-DH-N′-JH and VL-N-JL, which are individual to that cell. This means that the individual B cells differ from each other through differently rearranged IgH genes. This diversity is termed “polyclonality.” |
| •The tumor cells of B-cell lymphomas, in contrast, possess identical VH-N-DH-N′-JH and VL-N-JL sequences, indicating that they have arisen from the same transformed B-cell and, thereby, have formed a clone. This is termed “monoclonality”. |
| Rearrangement of the Immunoglobulin heavy chain (IgH) gene |
| • The first step occurs in the “early pre-B-cells” and involves the recombination of the D-(Diversity) and J-(Joining) segments of the heavy chain genes (IgH). During this process, one of 27 possible D-segments and one of 6 possible J-segments are randomly chosen and fused closely together, resulting in exclusion of the DNA segments which previously separated them (see Figure 2). In addition, nucleotides of variable length and composition are randomly inserted between the rearranged D- and J-segments (N′-region). |
| • The second step occurs in the “late pre-B-cells” and involves the rearrangement of the V-(Variable) segment, resulting in the complete rearrangement of the IgH gene (Figure 2). It involves the insertion of one of a possible 59 V-segments with the above-formed D-J-segment and the exclusion of the DNA segments which previously separated them (see Figure 2). In addition, as above, nucleotides of variable length and composition are randomly inserted between the rearranged D-J- segments and the V-segment (N-region). In this way, a different DNA segment (V-N-D-N′-J) for each B-cell is created, which serves as a “finger-print” for it and all of its' daughter cells. |
| Rearrangement of the Immunoglobulin light chain (IgL) gene |
| •The rearrangement of the two IgL-genes (κ and λ) also takes place in the pre-B-cell, and occurs in a similar manner to the rearrangement of IgH. In contrast to the IgH, the IgL-genes do not possess any D-segments. As a consequence the rearrangement of the IgL involves only one step, whereby nucleotides of variable length and composition (N-sequence) are inserted between the V- and J-segments. In this way, a second DNA segment (V-N-J) is created, which differs between each B-cell. When the Ig-gene rearrangements are completed the immature B cells have evolved. |
| Non-functional Ig-rearrangements |
| • Through the above-mentioned randomised insertion of nucleotides during the IgH and IgL rearrangements, “Stop-codons” or “Frame-shifts,” which inhibit Ig-protein functioning, occur relatively frequently. In such cases, a second independent rearrangement of either the IgH or IgL occurs. Should this additional rearrangement result in a non-coding DNA sequence and, therefore, to a non-functional Ig receptor molecule, the B-cell is eliminated via apoptosis. |
| Rearrangement of the Ig genes in normal and lymphomatous B-cells |
| • As described above, every cell which arises from progenitor B-cells possesses two Ig gene rearrangement products, VH-N-DH-N′-JH and VL-N-JL, which are individual to that cell. This means that the individual B cells differ from each other through differently rearranged IgH genes. This diversity is termed “polyclonality.” |
| •The tumor cells of B-cell lymphomas, in contrast, possess identical VH-N-DH-N′-JH and VL-N-JL sequences, indicating that they have arisen from the same transformed B-cell and, thereby, have formed a clone. This is termed “monoclonality”. |
Function of the germinal center reaction.
| • Generation of memory B cells and plasma cells. |
| • Affinity maturation of the Ig receptors by somatic hypermutation and antigen selection. |
| • Increase of the defence efficiency of the secreted Ig (antibodies) by changing their effector domains by means of isotype switch. |
| • Generation of memory B cells and plasma cells. |
| • Affinity maturation of the Ig receptors by somatic hypermutation and antigen selection. |
| • Increase of the defence efficiency of the secreted Ig (antibodies) by changing their effector domains by means of isotype switch. |
Chromosomal translocations of non-Hodgkin's lymphoma (NHL).
| NHL histologic type | Translocation | % of cases affected | Proto-oncogene involved | Mechanism of proto-oncogene activation | Proto-oncogene function |
|---|---|---|---|---|---|
| *in the adult population; 85% in childhood. | |||||
| Lymphoplasmacytic lymphoma | t(9;14)(p13;q32) | 50% | PAX-5 | Transcriptional deregulation | Transcription factor regulating B-cell proliferation and differentiation |
| Follicular lymphoma | t(14;18)(q32;q21) t(2;18)(p11;q21) t(18;22)(q21;q11) | 90% | BCL-2 | Transcriptional deregulation | Negative regulator of apoptosis |
| Mantle cell lymphoma | t(11;14)(q13;q32) | 70% | BCL-1/ cyclin D1 | Transcriptional deregulation | Cell cycle regulator |
| MALT lymphoma | t(11;18)(q21;q21) t(1;14)(p22;q32) | 50% rare | API2/MLT BCL-10 | Fusion protein Trancriptional deregulation | API2 has antiapoptotic activity Anti-apoptosis (?) |
| Diffuse large B-cell lymphoma | der(3)(q27) | 35% | BCL-6 | Transcriptional deregulation | Transcriptional repressor required for GC formation |
| Burkitt lymphoma | t(8;14)(q24;q32) t(2;8)(p11;q24) t(8;22)(q24;q11) | 80% 15% 5% | c-MYC | Transcriptional deregulation | Transcription factor regulating cell proliferation and growth |
| Anaplastic large T-cell lymphoma | t(2;5)(p23;q35) | 60%* | NPM/ALK | Fusion protein | ALK is a tyrosine kinase |
| NHL histologic type | Translocation | % of cases affected | Proto-oncogene involved | Mechanism of proto-oncogene activation | Proto-oncogene function |
|---|---|---|---|---|---|
| *in the adult population; 85% in childhood. | |||||
| Lymphoplasmacytic lymphoma | t(9;14)(p13;q32) | 50% | PAX-5 | Transcriptional deregulation | Transcription factor regulating B-cell proliferation and differentiation |
| Follicular lymphoma | t(14;18)(q32;q21) t(2;18)(p11;q21) t(18;22)(q21;q11) | 90% | BCL-2 | Transcriptional deregulation | Negative regulator of apoptosis |
| Mantle cell lymphoma | t(11;14)(q13;q32) | 70% | BCL-1/ cyclin D1 | Transcriptional deregulation | Cell cycle regulator |
| MALT lymphoma | t(11;18)(q21;q21) t(1;14)(p22;q32) | 50% rare | API2/MLT BCL-10 | Fusion protein Trancriptional deregulation | API2 has antiapoptotic activity Anti-apoptosis (?) |
| Diffuse large B-cell lymphoma | der(3)(q27) | 35% | BCL-6 | Transcriptional deregulation | Transcriptional repressor required for GC formation |
| Burkitt lymphoma | t(8;14)(q24;q32) t(2;8)(p11;q24) t(8;22)(q24;q11) | 80% 15% 5% | c-MYC | Transcriptional deregulation | Transcription factor regulating cell proliferation and growth |
| Anaplastic large T-cell lymphoma | t(2;5)(p23;q35) | 60%* | NPM/ALK | Fusion protein | ALK is a tyrosine kinase |
| **B and T/NK-cell neoplasms are grouped according to major clinical presentations (predominantly disseminated/leukemic, primary extranodal, predominantly nodal) |
| B-cell Neoplasms |
| Precursor B-cell neoplasm |
| • Precursor B-lymphoblastic leukemia/lymphoma (precursor B-cell acute lymphoblastic leukemia) |
| Mature (peripheral) B-cell neoplasms** |
| • Chronic lymphocytic leukemia/B-cell small lymphocytic lymphoma |
| • B-cell prolymphocytic leukemia |
| • Lymphoplasmacytic lymphoma |
| • Splenic marginal zone B-cell lymphoma (splenic lymphoma with villous lymphocytes) |
| • Hairy cell leukemia |
| • Plasma cell myeloma/Plasmacytoma |
| • Extranodal marginal zone B-cell lymphoma (MALT lymphoma) |
| • Nodal marginal zone B-cell lymphoma |
| • Follicular lymphoma |
| • Mantle cell lymphoma |
| • Diffuse large B-cell lymphomas |
| • Burkitt lymphoma/leukemia |
| T and NK-Cell Neoplasms |
| Precursor T-cell neoplasm |
| • Precursor T-lymphoblastic leukemia/lymphoma (precursor T-cell acute lymphoblastic leukemia) |
| • Blastoid NK-cell lymphoma |
| Mature (peripheral) T-cell neoplasms |
| • T-cell prolymphocytic leukemia |
| • T-cell large granular lymphocytic leukemia |
| • Aggressive NK-cell leukemia |
| • Adult T-cell lymphoma/leukemia (HTLV1+) |
| • Extranodal NK/T-cell lymphoma, nasal type |
| • Enteropathy-type T-cell lymphoma |
| • Hepatosplenic T-cell lymphoma |
| • Subcutaneous panniculitis-like T-cell lymphoma |
| • Mycosis fungoides/Sézary syndrome |
| • Primary cutaneous anaplastic large cell lymphoma |
| • Peripheral T-cell lymphoma, not otherwise specified |
| • Angioimmunoblastic T-cell lymphoma |
| • Primary systemic anaplastic large cell lymphoma |
| Hodgkin lymphoma (Hodgkin disease) |
| • Nodular lymphocyte predominant Hodgkin lymphoma |
| • Classical hodgkin lymphoma |
| Nodular sclerosis Hodgkin lymphoma (Grades 1 and 2) |
| Lymphocyte-rich classical Hodgkin lymphoma |
| Mixed cellularity Hodgkin lymphoma |
| Lymphocyte depleted Hodgkin lymphoma |
| **B and T/NK-cell neoplasms are grouped according to major clinical presentations (predominantly disseminated/leukemic, primary extranodal, predominantly nodal) |
| B-cell Neoplasms |
| Precursor B-cell neoplasm |
| • Precursor B-lymphoblastic leukemia/lymphoma (precursor B-cell acute lymphoblastic leukemia) |
| Mature (peripheral) B-cell neoplasms** |
| • Chronic lymphocytic leukemia/B-cell small lymphocytic lymphoma |
| • B-cell prolymphocytic leukemia |
| • Lymphoplasmacytic lymphoma |
| • Splenic marginal zone B-cell lymphoma (splenic lymphoma with villous lymphocytes) |
| • Hairy cell leukemia |
| • Plasma cell myeloma/Plasmacytoma |
| • Extranodal marginal zone B-cell lymphoma (MALT lymphoma) |
| • Nodal marginal zone B-cell lymphoma |
| • Follicular lymphoma |
| • Mantle cell lymphoma |
| • Diffuse large B-cell lymphomas |
| • Burkitt lymphoma/leukemia |
| T and NK-Cell Neoplasms |
| Precursor T-cell neoplasm |
| • Precursor T-lymphoblastic leukemia/lymphoma (precursor T-cell acute lymphoblastic leukemia) |
| • Blastoid NK-cell lymphoma |
| Mature (peripheral) T-cell neoplasms |
| • T-cell prolymphocytic leukemia |
| • T-cell large granular lymphocytic leukemia |
| • Aggressive NK-cell leukemia |
| • Adult T-cell lymphoma/leukemia (HTLV1+) |
| • Extranodal NK/T-cell lymphoma, nasal type |
| • Enteropathy-type T-cell lymphoma |
| • Hepatosplenic T-cell lymphoma |
| • Subcutaneous panniculitis-like T-cell lymphoma |
| • Mycosis fungoides/Sézary syndrome |
| • Primary cutaneous anaplastic large cell lymphoma |
| • Peripheral T-cell lymphoma, not otherwise specified |
| • Angioimmunoblastic T-cell lymphoma |
| • Primary systemic anaplastic large cell lymphoma |
| Hodgkin lymphoma (Hodgkin disease) |
| • Nodular lymphocyte predominant Hodgkin lymphoma |
| • Classical hodgkin lymphoma |
| Nodular sclerosis Hodgkin lymphoma (Grades 1 and 2) |
| Lymphocyte-rich classical Hodgkin lymphoma |
| Mixed cellularity Hodgkin lymphoma |
| Lymphocyte depleted Hodgkin lymphoma |
Frequency of B and T-cell neoplasms in the REAL classification. Data from reference 7.
| Diagnosis | % of Total Cases |
|---|---|
| Diffuse Large B-cell lymphoma | 31% |
| Follicular Lymphoma | 22% |
| MALT lymphoma | 8% |
| Mature T-cell lymphomas (except anaplastic large cell lymphoma) | 8% |
| Chronic lymphocytic leukemia/small lymphocytic lymphoma | 7% |
| Mantle Cell Lymphoma | 6% |
| Mediastinal Large B-cell Lymphoma | 2% |
| Anaplastic Large Cell Lymphoma | 2% |
| Burkitt Lymphoma | 2% |
| Nodal Marginal Zone Lymphoma | 2% |
| Precursor T Lymphoblastic | 2% |
| Lymphoplasmacytic Lymphoma | 1% |
| Other types | 7% |
| Diagnosis | % of Total Cases |
|---|---|
| Diffuse Large B-cell lymphoma | 31% |
| Follicular Lymphoma | 22% |
| MALT lymphoma | 8% |
| Mature T-cell lymphomas (except anaplastic large cell lymphoma) | 8% |
| Chronic lymphocytic leukemia/small lymphocytic lymphoma | 7% |
| Mantle Cell Lymphoma | 6% |
| Mediastinal Large B-cell Lymphoma | 2% |
| Anaplastic Large Cell Lymphoma | 2% |
| Burkitt Lymphoma | 2% |
| Nodal Marginal Zone Lymphoma | 2% |
| Precursor T Lymphoblastic | 2% |
| Lymphoplasmacytic Lymphoma | 1% |
| Other types | 7% |
Immunophenotypic and genetic features of common B-cell neoplasms
| Neoplasm | SIg; cIg | CD5 | CD10 | Bcl6 | CD23 | CD43 | CD103 | Cyclin D1 | CD 138 | Genetic Abnormality | Immunoglobulin Genes* |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: R, rearranged; M, mutated; NK, natural killer cell; U, unmutated; O, ongoing mutations; NA, not available | |||||||||||
| Key: + = >90% positive; +/− = > 50% positive; −/+ = < 50% positive; − = < 10% positive | |||||||||||
| B-SLL/CLL | +; −/+ | + | − | − | + | + | − | − | − | trisomy 12; 13q | R,U (50%); M (50%) |
| Lymphoplasmacytic lymphoma | +; + | − | − | − | − | +/− | − | − | −/+ | t(9;14); del 6(q23) | R,M |
| Hairy cell leukemia | +;− | − | − | − | − | + | ++ | +/− | − | none known | R,M |
| Plasma cell myeloma | −;+ | − | −/+ | − | − | −/+ | − | −/+ | + | t(4:14), t(6;14) t(14;16), t(1;14) | R,M |
| Splenic marginal zone lymphoma | +; −/+ | − | − | − | − | − | + | − | − | none known | R,M |
| Follicular lymphoma | +; − | − | +/− | + | −/+ | − | − | − | − | t(14;18); bcl-2 | R,M,O |
| Mantle cell lymphoma | +; − | + | − | − | − | + | − | + | − | t(11;14); bcl-1 | R,U |
| MALT lymphoma | +; +/− | − | − | − | −/+ | −/+ | − | − | − | +3, t(11;18); API2/MLT1 | R,M,O |
| Diffuse large B-cell lymphoma | +/−; −/+ | − | −/+ | +/− | NA | −/+ | NA | − | −/+ | t(14;18),t(8;14) 3q27; BCL2, cMYC, BCL6 | R,M |
| Burkitt lymphoma | +; − | − | + | + | − | − | NA | − | − | t(8;14), t(2;8), t(8;22); cMYC; EBV−/+ | R,M |
| Neoplasm | SIg; cIg | CD5 | CD10 | Bcl6 | CD23 | CD43 | CD103 | Cyclin D1 | CD 138 | Genetic Abnormality | Immunoglobulin Genes* |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: R, rearranged; M, mutated; NK, natural killer cell; U, unmutated; O, ongoing mutations; NA, not available | |||||||||||
| Key: + = >90% positive; +/− = > 50% positive; −/+ = < 50% positive; − = < 10% positive | |||||||||||
| B-SLL/CLL | +; −/+ | + | − | − | + | + | − | − | − | trisomy 12; 13q | R,U (50%); M (50%) |
| Lymphoplasmacytic lymphoma | +; + | − | − | − | − | +/− | − | − | −/+ | t(9;14); del 6(q23) | R,M |
| Hairy cell leukemia | +;− | − | − | − | − | + | ++ | +/− | − | none known | R,M |
| Plasma cell myeloma | −;+ | − | −/+ | − | − | −/+ | − | −/+ | + | t(4:14), t(6;14) t(14;16), t(1;14) | R,M |
| Splenic marginal zone lymphoma | +; −/+ | − | − | − | − | − | + | − | − | none known | R,M |
| Follicular lymphoma | +; − | − | +/− | + | −/+ | − | − | − | − | t(14;18); bcl-2 | R,M,O |
| Mantle cell lymphoma | +; − | + | − | − | − | + | − | + | − | t(11;14); bcl-1 | R,U |
| MALT lymphoma | +; +/− | − | − | − | −/+ | −/+ | − | − | − | +3, t(11;18); API2/MLT1 | R,M,O |
| Diffuse large B-cell lymphoma | +/−; −/+ | − | −/+ | +/− | NA | −/+ | NA | − | −/+ | t(14;18),t(8;14) 3q27; BCL2, cMYC, BCL6 | R,M |
| Burkitt lymphoma | +; − | − | + | + | − | − | NA | − | − | t(8;14), t(2;8), t(8;22); cMYC; EBV−/+ | R,M |
Immunophenotypic and genetic features of common T-cell neoplasms.
| Neoplasm | CD3 (S;C) | CD5 | CD7 | CD4 | CD8 | CD30 | TCR | NK16, 56 | Cytotoxic granule | EBV | Genetic Abnormality | T-Receptor Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: R, rearranged; M, mutated; NK, natural killer cell; U, unmutated; O, ongoing mutations; TCR, T-cell receptor gene; Ig, immunoglobulin; NA, not available | ||||||||||||
| Key: + = >90% positive; +/− = > 50% positive; −/+ = < 50% positive; − = < 10% positive; Cytotoxic granule = TIA-1, perforin, and/or granzyme | ||||||||||||
| * Mutations in the Ig gene V region indicate exposure to antigen. | ||||||||||||
| T-prolymphocytic leukemia | + | − | +,+ | +/− | −/+ | − | αβ | − | − | − | inv 14 + 8 | R |
| T-large granular lymphoproliferative disease | + | − | +,+ | − | + | − | αβ | +,− | + | − | none known | R |
| NK large granlular lymphoproliferative disease | − | − | +, − | − | +/− | − | − | −,+ | + | + | none known | G |
| Extranodal NK/T-cell lymphoma | −;+ | − | −/+ | − | − | − | − | NA,+ | + | ++ | none known | G |
| Hepatosplenic T-cell lymphoma | + | − | + | − | − | − | γδ>>αβ | +,−/+ | + | − | i(7q)(q10) | R |
| Enteropathy-type T-cell lymphoma | + | + | + | − | +/− | +/− | αβ>>γδ | − | + | − | none known | R |
| Mycosis fungoides | + | + | −/+ | + | − | − | αβ | − | − | − | None known | R |
| Cutaneous anaplastic large cell lymphoma | + | +/− | +/− | +/− | − | ++ | αβ | − | −/+ | − | none known | R |
| Subcutaneous panniculitis-like T-cell | + | + | + | − | + | −/+ | αβ>γδ | −,+/− | + | − | none known | R |
| Peripheral T-cell lymphoma, unspecified | +/− | +/− | +/− | +/− | −/+ | −/+ | αβ>γδ | −/+ | −/+ | −/+ | inv 14; +8, complex | R |
| Angioimmunoblastic | + | + | + | +/− | −/+ | − | αβ | − | NA | +/− | +3 +5 +X | R |
| Primary systemic anaplastic large cell lymphoma | +/− | +/− | NA | −/+ | −/+ | ++ | αβ | − | + | − | t(2;5); NPM/ALK | R |
| Neoplasm | CD3 (S;C) | CD5 | CD7 | CD4 | CD8 | CD30 | TCR | NK16, 56 | Cytotoxic granule | EBV | Genetic Abnormality | T-Receptor Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: R, rearranged; M, mutated; NK, natural killer cell; U, unmutated; O, ongoing mutations; TCR, T-cell receptor gene; Ig, immunoglobulin; NA, not available | ||||||||||||
| Key: + = >90% positive; +/− = > 50% positive; −/+ = < 50% positive; − = < 10% positive; Cytotoxic granule = TIA-1, perforin, and/or granzyme | ||||||||||||
| * Mutations in the Ig gene V region indicate exposure to antigen. | ||||||||||||
| T-prolymphocytic leukemia | + | − | +,+ | +/− | −/+ | − | αβ | − | − | − | inv 14 + 8 | R |
| T-large granular lymphoproliferative disease | + | − | +,+ | − | + | − | αβ | +,− | + | − | none known | R |
| NK large granlular lymphoproliferative disease | − | − | +, − | − | +/− | − | − | −,+ | + | + | none known | G |
| Extranodal NK/T-cell lymphoma | −;+ | − | −/+ | − | − | − | − | NA,+ | + | ++ | none known | G |
| Hepatosplenic T-cell lymphoma | + | − | + | − | − | − | γδ>>αβ | +,−/+ | + | − | i(7q)(q10) | R |
| Enteropathy-type T-cell lymphoma | + | + | + | − | +/− | +/− | αβ>>γδ | − | + | − | none known | R |
| Mycosis fungoides | + | + | −/+ | + | − | − | αβ | − | − | − | None known | R |
| Cutaneous anaplastic large cell lymphoma | + | +/− | +/− | +/− | − | ++ | αβ | − | −/+ | − | none known | R |
| Subcutaneous panniculitis-like T-cell | + | + | + | − | + | −/+ | αβ>γδ | −,+/− | + | − | none known | R |
| Peripheral T-cell lymphoma, unspecified | +/− | +/− | +/− | +/− | −/+ | −/+ | αβ>γδ | −/+ | −/+ | −/+ | inv 14; +8, complex | R |
| Angioimmunoblastic | + | + | + | +/− | −/+ | − | αβ | − | NA | +/− | +3 +5 +X | R |
| Primary systemic anaplastic large cell lymphoma | +/− | +/− | NA | −/+ | −/+ | ++ | αβ | − | + | − | t(2;5); NPM/ALK | R |
Genetic abnormalities in lymphoid neoplasms associated with diagnostically useful protein expression.
| Translocation | Protein Expression | Distribution | Utility |
|---|---|---|---|
| t(14;18) | Bcl2 | Germinal centers − | Reactive vs neoplastic follicles |
| Follicular lymphoma + | |||
| t(11;14) | Cyclin D1 | Mantle cell lymphoma (all) | Mantle cell lymphoma vs other lymphomas or reactive process |
| Hairy cell leukemia (some) | |||
| Plasma cell myeloma (some) | |||
| t(2;5) | ALK (anaplastic lymphoma kinase) | Anaplastic large-cell lymphoma (most) | ALCL vs other lymphomas; prognosis of ALCL |
| t(1;14) or t(11;18) | Bcl10 | MALT lymphomas unresponsive to Helicobacter pylori eradication | Prognosis/response prediction of MALT lymphomas |
| Translocation | Protein Expression | Distribution | Utility |
|---|---|---|---|
| t(14;18) | Bcl2 | Germinal centers − | Reactive vs neoplastic follicles |
| Follicular lymphoma + | |||
| t(11;14) | Cyclin D1 | Mantle cell lymphoma (all) | Mantle cell lymphoma vs other lymphomas or reactive process |
| Hairy cell leukemia (some) | |||
| Plasma cell myeloma (some) | |||
| t(2;5) | ALK (anaplastic lymphoma kinase) | Anaplastic large-cell lymphoma (most) | ALCL vs other lymphomas; prognosis of ALCL |
| t(1;14) or t(11;18) | Bcl10 | MALT lymphomas unresponsive to Helicobacter pylori eradication | Prognosis/response prediction of MALT lymphomas |
Algorithm for using special techniques in lymphoma diagnosis.
| *Can be detected by gene expression (immunophenotype) or gene rearrangement (genetic techniques) |
| Abbreviations: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; MALT, mucosa associated lymphoid tissue; Ig, immunoglobulin; DLBCL, diffuse large B-cell lymphoma; NK, natural killer cell; TdT, terminal deoxynucleotidyl transferase; ALK, anaplastic lymphoma kinase. |
| A. Morphology: Small lymphoid cells |
| 1. Benign vs malignant |
| *Immunoglobulin light chains (kappa and lambda) |
| *Bcl2 protein in follicles |
| Kappa/lambda polyclonal or Bcl2- follicles =Reactive |
| Kappa/lambda monoclonal or Bcl2+ follicles = Small B-cell lymphoma |
| 2. Classification of Small B-cell Neoplasms |
| CD5+: CLL/SLL vs mantle cell lymphoma |
| *Cyclin D1-, dim sIg, CD23+ = CLL/SLL |
| *Cyclin D1+, bright sIg, CD23− = Mantle cell lymphoma |
| CD5-: Follicular lymphoma vs Marginal zone lymphoma (MALT) |
| CD10+ = Follicular lymphoma |
| CD10− = Marginal zone lymphoma vs CD10− follicular lymphoma |
| Bcl6+, CD43−, *Bcl2+ follicles = follicular lymphoma |
| Bcl6−, CD43+, Bcl2− follicles = marginal zone lymphoma |
| B. Morphology: Large lymphoid cells |
| 1. Lymphoma vs nonlymphoid tumor |
| CD45 (leukocyte common antigen), pan- B and pan-T-cell antigens,* immunoglobulin light chains, cytokeratin, melanoma markers |
| 2. Subclassification of medium-sized and large cell lymphomas |
| Pan-B antigens (CD20, CD79a)+ = B-cell lymphoma |
| Ig+ = mature B-cell lymphoma (DLBCL vs Burkitt) |
| CD10+ bcl2- Ki67 >99% = favors Burkitt |
| CD10− bcl2+ Ki67 <90% = favors DLBCL |
| Ig- = mature B-cell vs lymphoblastic |
| TdT+ CD10+ = precursor B-lymphoblastic |
| TdT− CD10−/+ =DLBCL |
| Pan-T antigens + (CD3, CD2, CD45RO) = T/NK-cell lymphoma |
| TdT+ = precursor T-lymphoblastic |
| TdT− = mature T/NK-cell lymphoma |
| CD21+ FDC, CD10+ = angioimmunoblastic T-cell lymphoma |
| CD30+ ALK+ = anaplastic large cell lymphoma |
| *Can be detected by gene expression (immunophenotype) or gene rearrangement (genetic techniques) |
| Abbreviations: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; MALT, mucosa associated lymphoid tissue; Ig, immunoglobulin; DLBCL, diffuse large B-cell lymphoma; NK, natural killer cell; TdT, terminal deoxynucleotidyl transferase; ALK, anaplastic lymphoma kinase. |
| A. Morphology: Small lymphoid cells |
| 1. Benign vs malignant |
| *Immunoglobulin light chains (kappa and lambda) |
| *Bcl2 protein in follicles |
| Kappa/lambda polyclonal or Bcl2- follicles =Reactive |
| Kappa/lambda monoclonal or Bcl2+ follicles = Small B-cell lymphoma |
| 2. Classification of Small B-cell Neoplasms |
| CD5+: CLL/SLL vs mantle cell lymphoma |
| *Cyclin D1-, dim sIg, CD23+ = CLL/SLL |
| *Cyclin D1+, bright sIg, CD23− = Mantle cell lymphoma |
| CD5-: Follicular lymphoma vs Marginal zone lymphoma (MALT) |
| CD10+ = Follicular lymphoma |
| CD10− = Marginal zone lymphoma vs CD10− follicular lymphoma |
| Bcl6+, CD43−, *Bcl2+ follicles = follicular lymphoma |
| Bcl6−, CD43+, Bcl2− follicles = marginal zone lymphoma |
| B. Morphology: Large lymphoid cells |
| 1. Lymphoma vs nonlymphoid tumor |
| CD45 (leukocyte common antigen), pan- B and pan-T-cell antigens,* immunoglobulin light chains, cytokeratin, melanoma markers |
| 2. Subclassification of medium-sized and large cell lymphomas |
| Pan-B antigens (CD20, CD79a)+ = B-cell lymphoma |
| Ig+ = mature B-cell lymphoma (DLBCL vs Burkitt) |
| CD10+ bcl2- Ki67 >99% = favors Burkitt |
| CD10− bcl2+ Ki67 <90% = favors DLBCL |
| Ig- = mature B-cell vs lymphoblastic |
| TdT+ CD10+ = precursor B-lymphoblastic |
| TdT− CD10−/+ =DLBCL |
| Pan-T antigens + (CD3, CD2, CD45RO) = T/NK-cell lymphoma |
| TdT+ = precursor T-lymphoblastic |
| TdT− = mature T/NK-cell lymphoma |
| CD21+ FDC, CD10+ = angioimmunoblastic T-cell lymphoma |
| CD30+ ALK+ = anaplastic large cell lymphoma |

Events in B-cell development.
The development and maturation process of B cells begins in the bone marrow. Here, the “pre-B cell” arises from the “progenitor (Pro) B cell” following rearrangement of the immunoglobulin heavy chain gene (symbolized with horizontal lines in black). Subsequently, rearrangement of the light chain genes occurs resulting in the expression of the whole immunoglobulin molecule on the cell surface, serving as an antigen receptor. With the production of this “immature” B cell, the initial phase of B-cell development is, thereby, completed. The “immature” B cell is so defined since it is unable to initiate an immune response following the presentation of a foreign antigen. The B-cell attains this ability only on leaving the bone marrow, passing through the blood stream and entering the peripheral lymphoid tissue. Here, the B cell migrates to the outer region of the lymph node in the “primary” follicles and, later, to the follicle mantles. This differentiation step is associated with the additional expression of IgD. These IgM+/IgD+ B cells are known as “naive mature B-cells”. When these cells come into contact with antigen (AG), which can bind to their immunoglobulin molecules, they transform into proliferating extrafollicular B blasts, from which short-lived plasma cells and “antigen-induced” or “primed” B cells are derived. These “primed” B cells initiate and maintain the germinal center reaction, during which they transform into rapidly proliferating centroblasts. During the mitotic proliferation and differentiation of the centroblasts into centrocytes, somatic mutations in the variable region of the immunoglobulin genes are inserted in a randomized manner (the mutations are represented by vertical lines). The centrocytes with advantageous mutations (i.e. those which lead to an increase in the affinity of the immunoglobulin receptor) differentiate further, passing out of the germinal centre into long-lived plasma cells or into “memory” B cells. The latter remain in the marginal zone. FDC, folicular dendritic cell;  , apoptosis
, apoptosis
As a result of the differentiation phases of B-cells and of the somatic mutation process, 3 major different mature forms of B-cells can be identified:
• Naive mature B-cells (recirculating and sessile subtypes)
• Germinal center B-cells (centroblasts and centrocytes)
• Post germinal center B-cells which include memory B cells and long-lived plasma cells
From all of these different B-cell forms, malignant B-cell lymphomas arise, which distinguish themselves clinically and which are characterized in their biological behavior not only by the transformation event but also by the inherent characteristics of the cell of origin. Classical Hodgkin lymphomas, in which the phenotypical and clinical features are predominantly determined by the transformation event, are an exception to this rule.
Events in B-cell development.
The development and maturation process of B cells begins in the bone marrow. Here, the “pre-B cell” arises from the “progenitor (Pro) B cell” following rearrangement of the immunoglobulin heavy chain gene (symbolized with horizontal lines in black). Subsequently, rearrangement of the light chain genes occurs resulting in the expression of the whole immunoglobulin molecule on the cell surface, serving as an antigen receptor. With the production of this “immature” B cell, the initial phase of B-cell development is, thereby, completed. The “immature” B cell is so defined since it is unable to initiate an immune response following the presentation of a foreign antigen. The B-cell attains this ability only on leaving the bone marrow, passing through the blood stream and entering the peripheral lymphoid tissue. Here, the B cell migrates to the outer region of the lymph node in the “primary” follicles and, later, to the follicle mantles. This differentiation step is associated with the additional expression of IgD. These IgM+/IgD+ B cells are known as “naive mature B-cells”. When these cells come into contact with antigen (AG), which can bind to their immunoglobulin molecules, they transform into proliferating extrafollicular B blasts, from which short-lived plasma cells and “antigen-induced” or “primed” B cells are derived. These “primed” B cells initiate and maintain the germinal center reaction, during which they transform into rapidly proliferating centroblasts. During the mitotic proliferation and differentiation of the centroblasts into centrocytes, somatic mutations in the variable region of the immunoglobulin genes are inserted in a randomized manner (the mutations are represented by vertical lines). The centrocytes with advantageous mutations (i.e. those which lead to an increase in the affinity of the immunoglobulin receptor) differentiate further, passing out of the germinal centre into long-lived plasma cells or into “memory” B cells. The latter remain in the marginal zone. FDC, folicular dendritic cell; , apoptosis
As a result of the differentiation phases of B-cells and of the somatic mutation process, 3 major different mature forms of B-cells can be identified:
• Naive mature B-cells (recirculating and sessile subtypes)
• Germinal center B-cells (centroblasts and centrocytes)
• Post germinal center B-cells which include memory B cells and long-lived plasma cells
From all of these different B-cell forms, malignant B-cell lymphomas arise, which distinguish themselves clinically and which are characterized in their biological behavior not only by the transformation event but also by the inherent characteristics of the cell of origin. Classical Hodgkin lymphomas, in which the phenotypical and clinical features are predominantly determined by the transformation event, are an exception to this rule.

Model of B-cell non-Hodgkin's lymphoma (NHL) histogenesis and pathogenesis.
A lymphoid follicle, constituted by the germinal center (GC) and the mantle zone (MZ), is represented together with the surrounding marginal zone (MargZ). Based on the absence or presence of IgV somatic mutations, B-cell NHL can be distinguished into two broad histogenetic categories: i) pre-GC derived NHL, lacking IgV mutations and including mantle cell lymphoma (MCL); ii) B-cell NHL derived from a cell that transited through the GC and harboring mutated IgV genes, exemplified in the figure by follicular lymphoma (FL), lymphoplasmacytic lymphoma (LPL), MALT lymphoma, diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). In B-cell chronic lymphocytic leukemia/ small lymphocytic lymphoma (CLL/SLL), the presence of somatically mutated IgV genes in >50% of the cases also suggests a derivation from a GC experienced B cell.49 For each category, the arrow indicating the histogenetic origin is flanked by the genetic lesion most frequently associated with the lymphoma. In CLL/SLL, as well as in a subset of DLBCL, the relevant cancer related gene has not been identified.
Model of B-cell non-Hodgkin's lymphoma (NHL) histogenesis and pathogenesis.
A lymphoid follicle, constituted by the germinal center (GC) and the mantle zone (MZ), is represented together with the surrounding marginal zone (MargZ). Based on the absence or presence of IgV somatic mutations, B-cell NHL can be distinguished into two broad histogenetic categories: i) pre-GC derived NHL, lacking IgV mutations and including mantle cell lymphoma (MCL); ii) B-cell NHL derived from a cell that transited through the GC and harboring mutated IgV genes, exemplified in the figure by follicular lymphoma (FL), lymphoplasmacytic lymphoma (LPL), MALT lymphoma, diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). In B-cell chronic lymphocytic leukemia/ small lymphocytic lymphoma (CLL/SLL), the presence of somatically mutated IgV genes in >50% of the cases also suggests a derivation from a GC experienced B cell.49 For each category, the arrow indicating the histogenetic origin is flanked by the genetic lesion most frequently associated with the lymphoma. In CLL/SLL, as well as in a subset of DLBCL, the relevant cancer related gene has not been identified.


Chromosomal translocations and somatic hypermutation of the BCL-6 5′ noncoding region in diffuse large B cell lymphoma (DLBCL).
Chromosomal translocations and somatic hypermutation of the BCL-6 5′ noncoding region in diffuse large B cell lymphoma (DLBCL).

Process of agglomerative (bottom-up, clumping) hierarchical clustering.
The figure is a diagrammatic representation of the process of agglomerative hierarchical clustering. Each dot represents a sample associated with a certain gene expression pattern. The computer algorithm will identify pairs of samples with the most similarity in expression pattern. Samples are added to these original pairs based now on the average pattern of gene expression of each of the pairs. This process is repeated, giving rise to larger and larger clusters and eventually all the samples will be grouped together under one cluster. The cluster can be represented in the form of a dendrogram as illustrated in Figure 8 (color page 545).
Process of agglomerative (bottom-up, clumping) hierarchical clustering.
The figure is a diagrammatic representation of the process of agglomerative hierarchical clustering. Each dot represents a sample associated with a certain gene expression pattern. The computer algorithm will identify pairs of samples with the most similarity in expression pattern. Samples are added to these original pairs based now on the average pattern of gene expression of each of the pairs. This process is repeated, giving rise to larger and larger clusters and eventually all the samples will be grouped together under one cluster. The cluster can be represented in the form of a dendrogram as illustrated in Figure 8 (color page 545).
