
Abstract
Hemophilia A and B are monogenic disorders that were felt to be ideal targets for initiation of gene therapy. Although the first hemophilia gene therapy trial has been over 10 years ago, few trials are currently actively recruiting. Although preclinical studies in animals were promising, levels achieved in humans did not achieve long-term expression at adequate levels to achieve cures. Transplantation as a source of cellular replacement therapy for both hemophilia A and B have been successful following liver transplantation in which the recipient produces normal levels of either factor VIII (FVIII) or factor IX (FIX). Most of these transplants have been conducted for the treatment of liver failure rather than for “curing” hemophilia. There are a variety of new strategies for delivering the missing clotting factor through ectopic expression of the deficient protein. One approach uses hematopoietic stem cells using either a nonspecific promoter or using a lineage-specific promoter. An alternative strategy includes enhanced expression in endothelial cells or blood-outgrowth endothelial cells. An additional approach includes the expression of FVIII or FIX intraarticularly to mitigate the intraarticular bleeding that causes much of the disability for hemophilia patients. Because activated factor VII (FVIIa) can be used to treat patients with inhibitory antibodies to replacement clotting factors, preclinical gene therapy has been performed using platelet- or liver-targeted FVIIa expression. All of these newer approaches are just beginning to be explored in large animal models. Whereas improved recombinant replacement products continue to be the hallmark of hemophilia therapy, the frequency of replacement therapy is beginning to be addressed through longer-acting replacement products. A safe cure of hemophilia is still the desired goal, but many barriers must still be overcome.
Gene therapy of hemophilia has been the goal for treatment of hemophilia A or B, since the initial cloning of the genes more than 20 years ago. Cloning of the genes revolutionized the therapy of both hemophilia A and B. A high proportion of therapy today utilizes recombinant therapeutic factor concentrates. Initial development of recombinant factor VIII (FVIII) was optimized by the coexpression of von Willebrand factor (VWF) that optimized the intracellular trafficking and folding of FVIII. One therapeutic product utilized an expression system that produced FVIII from which the B domain had essentially been removed. This is important because most FVIII expression vectors being studied for gene therapy have been similarly truncated to facilitate packaging of the vector construct for which full-length FVIII's size is often problematic. Most gene therapy strategies for hemophilia A do not express FVIII in the context of a cell-synthesizing VWF. In platelets and endothelial cells, coexpression of FVIII with VWF has been beneficial and improves levels of FVIII expression. Recombinant factor IX (FIX), while effectively therapeutic, was found to have some variability in posttranslational processing. This affects the in vivo recovery, but not the plasma half-life. Differences in FIX posttranslational processing has also been seen in vivo following myotube-synthesized FIX. In clinical and preclinical studies, FVIII or FIX has been expressed sometimes in tissues that have not been demonstrated to normally synthesize and process these proteins physiologically. For both FIX and FVIII, their only carefully characterized function is in plasma; thus, ectopic expression may not be problematic, but the expressed protein must be studied carefully to ensure structure, function, and nonimmunogenicity. Recombinant activated factor VII (FVIIa) has been used for years to treat hemophilia patients1 who have developed inhibitory antibodies to the deficient clotting factor, although its half-life is short and there are some concerns about thrombogenicity.2
Previous Human Gene Therapy Clinical Trials for Hemophilia
The first gene therapy trial for FVIII deficiency involved nonviral somatic cell gene therapy using autologous fibroblasts (obtained from a skin biopsy) transfected with a plasmid containing the FVIII cDNA for B domain-deleted FVIII.3 Cells in culture that expressed high levels of FVIII were selected, expanded, characterized, and then implanted in the omentum at several sites using laparoscopy. Although FVIII levels of 1 to 5 U/dL were observed following implantation of the transfected fibroblast, none of the subjects had sustained plasma FVIII levels > 0.5 U/dL at 12 months. There was some decrease in use of replacement FVIII products, but further studies were not reported.
The first gene therapy trial for hemophilia B utilized adeno-associated virus (AAV)-mediated FIX gene transfer to skeletal muscle by direct injection.4,5 Circulating plasma levels were < 2% of normal in all individuals, with most being < 1% of normal. The limitation of therapeutic efficacy has prompted alternative strategies to direct muscle delivery most recently with intravascular delivery along with immunosuppression in hemophilia B dogs.6
Another clinical trial was conducted using a single portal infusion of AAV-2 vector expressing human FIX.7 Although vector infusion was not associated with acute or long-lasting toxicity, the transduced hepatocytes were targeted by cell-mediated immunity caused by the AAV capsids. This resulted in a decline in FIX and a transient transaminitis. The authors concluded that future studies may require immunomodulation to achieve long-term expression.
A phase I study of FVIII gene therapy utilizing a retroviral vector carrying the B domain-deleted FVIII gene was performed on 13 individuals.8 Vector sequences were detected in peripheral blood mononuclear cells for as long as 53 weeks posttreatment. Plasma FVIII levels in most recipients were minimally detectable, but the publication primarily emphasized a reduction in bleeding frequency and an improved area under the curve following FVIII infusion. An improved area under the curve does not seem to be a strong endpoint. A major reduction in frequency of therapeutic FVIII infusions was only seen in 1 of 12 subjects. Further studies with this approach were not conducted.
Although FVIII gene therapy preclinical studies have usually utilized B domain-deleted FVIII constructs, one clinical trial has used a full-length FVIII construct that was placed into a “gutted” adenoviral vector. Only one patient was treated, and because of thrombocytopenia and transaminitis, no additional subjects were enrolled in the trial; and we only have secondhand discussion of the results in the literature.9
Current Hemophilia Gene Therapy Trials
As of this writing, there are only three active hemophilia gene therapy trials. Two trials using FIX AAV vectors for hemophilia B are enrolling subjects. The first is a phase I study of AAV2-hFIX with portal artery injection, and the other is a phase I/II study using a modified AAV vector. The third active clinical trial is a phase IIa study that uses an oral agent, PTC 124, that in animals has demonstrated the ability to read through stop codons. Currently, no published information is available concerning these human clinical trials.
Gene Therapy Directed at the Local Delivery of Clotting Factor
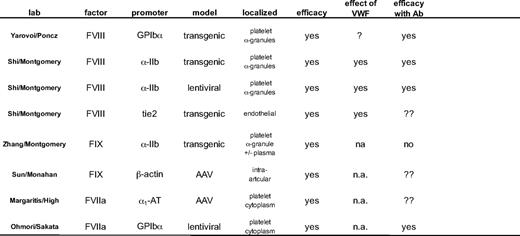
Several new approaches have been developed to deliver the missing clotting factor locally through intraarticular, platelet, or endothelial cell delivery and are summarized in Table 1. Whereas most of these approaches have resulted in long-term therapeutic correcting, the risk of insertional mutagenesis must be addressed.10
Intraarticular Gene Therapy of Hemophilia
Because joint bleeding is probably the most major chronic morbidity in hemophilia, one approach has utilized AAV-FIX administered directly into the knees of FIX knockout (KO) mice.11 After 4 weeks of AAV-FIX expression, joint bleeds were induced by needle puncture. Synovitis was observed in 100% of KO mice and was significantly reduced in joints receiving local intraarticular gene therapy. Whether this approach is effective in the presence of FIX inhibitors is not yet known.
Platelet-Directed Expression of FVIII
Two groups have focused on the delivery of FVIII by inducing expression in platelets. One group utilizes the glycoprotein Ibα promoter to drive FVIII expression in platelets,12 and our group uses the αIIb promoter to drive FVIII expression.13,14
FVIII is neither synthesized nor stored physiologically in megakaryocytes or platelets, yet VWF has been known to bind FVIII and serve as its carrier protein in plasma for many years. The initial synthesis of recombinant FVIII required the coexpression of VWF to optimize FVIII expression by cell lines producing recombinant FVIII, a process that is still utilized today in the commercial production of some recombinant FVIII.15 Several strategies were pursued by our group to demonstrate the efficacy of FVIII expressed in megakaryocytes. Transgenic mice were developed in which the FVIII gene was placed under the control of the αIIb promoter (here termed 2bF8), and then the transgene was bred onto the FVIII KO background.13,14 Secondly, a lentiviral vector was developed using a similar construct. Murine or human bone marrow hematopoietic stem cells (HSCs) were transduced with LV-2bF8. Figure 1 demonstrates that megakaryocytes derived from these transduced HSC produced FVIII, and this FVIII was stored together with VWF in megakaryocytes.16 In separate experiments, this FVIII was stored with VWF in α-granules (immunofluorescence or electron microscopy). Both proteins were released together by platelet agonists. Even though plasma FVIII was undetectable in the 2bF8-transgenic mice (FVIII KO background), the transgene expressed in platelets such that the whole blood FVIII was approximately 1% of normal whole blood FVIII (Figure 2A). If the transgene was bred onto a double KO background (FVIII–/– VWF–/–), the amount of platelet FVIII was reduced in homozygous (VWF–/–) mice (Figure 2B), but FVIII was still expressed in the absence of VWF, even though the amount of FVIII was significantly reduced. Furthermore, not only was survival of mice with the transgene normalized after tail clip challenge, but also the transgene had efficacy even in the presence of FVIII inhibitory antibodies with titers of 250 Bethesda Units (BU/mL) (Figure 3). The rationale for this efficacy was two-fold. Intraplatelet-stored FVIII was protected from plasma inhibitory antibodies and, even when released at sites of vascular injury, FVIII inhibitory antibodies are not immediate-acting antibodies and in vitro require 1 to 2 hours of incubation to get a 50% reduction in FVIII activity—ample time for the released platelet FVIII to have therapeutic efficacy in vivo.

Induced FVIII expression in megakaryocytes under the control of the α-IIb promoter. Murine FVIII–/– bone marrow-derived hematopoietic stem cells were transduced with a lentiviral B domain-deleted cDNA construct for human FVIII (hFVIII) placed under the control of the integrin α-IIb promoter (2bF8) and the cells cultured in the presence of interleukin-3, interleukin-6, stem cell factor, thrombopoietin, and flt-3 ligand. Cells were immunostained for VWF (red) and FVIII (green). Untransduced cells only demonstrated VWF staining (A-D), whereas the 2bF8-transduced cells demonstrated FVIII immunostaining and colocalization of the FVIII with VWF (E-H and I-L). (Used with permission, with full details provided in J Thromb Haemostas. 2006;5:352-361.)
Induced FVIII expression in megakaryocytes under the control of the α-IIb promoter. Murine FVIII–/– bone marrow-derived hematopoietic stem cells were transduced with a lentiviral B domain-deleted cDNA construct for human FVIII (hFVIII) placed under the control of the integrin α-IIb promoter (2bF8) and the cells cultured in the presence of interleukin-3, interleukin-6, stem cell factor, thrombopoietin, and flt-3 ligand. Cells were immunostained for VWF (red) and FVIII (green). Untransduced cells only demonstrated VWF staining (A-D), whereas the 2bF8-transduced cells demonstrated FVIII immunostaining and colocalization of the FVIII with VWF (E-H and I-L). (Used with permission, with full details provided in J Thromb Haemostas. 2006;5:352-361.)

FVIII in plasma and platelets in 2bF8-transgenic mice. Transgenic mice were developed with the cDNA for B domain-deleted FVIII placed under the control of the integrin α-IIb promoter and bred onto the murine FVIIInull background. These transgenic mice (2bF8) only expressed FVIII in megakaryocytes and stored FVIII together with VWF in platelets. (A) Plasma FVIII was measured and was present in wild-type (WT), but was absent in plasma from either FVIIInull or 2bF8 transgenic (FVIIInull) mice. (B) Platelet FVIII was measured in murine platelets and was absent in wild-type and FVIIInull mice; but, in 2bF8 transgenic mice, platelet FVIII was present and was greater in homozygous transgenic mice (2bF8trans+/+) than in heterozygous (2bF8trans+/–) mice. To determine the effect of VWF on the expression of FVIII in platelets, the 2bF8 transgene was bred onto the double KO background (FVIIInull VWFnull). Platelet FVIII expression was reduced in the absence of VWF. Bone marrow transplantation (BMT) from a heterozygous 2bF8 mouse conferred similar expression levels when transplanted into FVIIInull mice following ablative bone marrow conditioning. In data not illustrated in this figure, platelet FVIII conferred survival following tail-clip challenge in contrast to the FVIIInull mice in which this challenge was uniformly fatal. *P < .001. (Used with permission, with full details provided in J Clin Invest. 2006;116:1974-1982.)
FVIII in plasma and platelets in 2bF8-transgenic mice. Transgenic mice were developed with the cDNA for B domain-deleted FVIII placed under the control of the integrin α-IIb promoter and bred onto the murine FVIIInull background. These transgenic mice (2bF8) only expressed FVIII in megakaryocytes and stored FVIII together with VWF in platelets. (A) Plasma FVIII was measured and was present in wild-type (WT), but was absent in plasma from either FVIIInull or 2bF8 transgenic (FVIIInull) mice. (B) Platelet FVIII was measured in murine platelets and was absent in wild-type and FVIIInull mice; but, in 2bF8 transgenic mice, platelet FVIII was present and was greater in homozygous transgenic mice (2bF8trans+/+) than in heterozygous (2bF8trans+/–) mice. To determine the effect of VWF on the expression of FVIII in platelets, the 2bF8 transgene was bred onto the double KO background (FVIIInull VWFnull). Platelet FVIII expression was reduced in the absence of VWF. Bone marrow transplantation (BMT) from a heterozygous 2bF8 mouse conferred similar expression levels when transplanted into FVIIInull mice following ablative bone marrow conditioning. In data not illustrated in this figure, platelet FVIII conferred survival following tail-clip challenge in contrast to the FVIIInull mice in which this challenge was uniformly fatal. *P < .001. (Used with permission, with full details provided in J Clin Invest. 2006;116:1974-1982.)

Platelet-induced expression of FVIII efficacious even in the presence of high-titer anti-FVIII inhibitory antibodies. (A) Recombinant human FVIII normalizes the survival of FVIIInull mice subjected to tail-clip survival when plasma FVIII levels are ≥ 2 IU/dL (no platelet FVIII). Mice with 2 IU/dL were infused with anti-FVIII inhibitory antibodies and survival to tail-clip challenge determined. Anti-FVIII inhibitory plasma abrogated the effect of 2 IU/dL of plasma FVIII with a reduction in survival (100% survival reduced to 33%) at 2.5 BU and no survival with higher titers (25 BU and 250 BU). (B) 2bF8-transgenic mice with no plasma FVIII (whole-blood FVIII level corresponding to 1.23% of normal) had normal survival, even in the presence of high-titer anti-FVIII inhibitory antibodies (250 BU). (Used with permission, with full details provided in J Clin Invest. 2006;116:1974-1982)
Platelet-induced expression of FVIII efficacious even in the presence of high-titer anti-FVIII inhibitory antibodies. (A) Recombinant human FVIII normalizes the survival of FVIIInull mice subjected to tail-clip survival when plasma FVIII levels are ≥ 2 IU/dL (no platelet FVIII). Mice with 2 IU/dL were infused with anti-FVIII inhibitory antibodies and survival to tail-clip challenge determined. Anti-FVIII inhibitory plasma abrogated the effect of 2 IU/dL of plasma FVIII with a reduction in survival (100% survival reduced to 33%) at 2.5 BU and no survival with higher titers (25 BU and 250 BU). (B) 2bF8-transgenic mice with no plasma FVIII (whole-blood FVIII level corresponding to 1.23% of normal) had normal survival, even in the presence of high-titer anti-FVIII inhibitory antibodies (250 BU). (Used with permission, with full details provided in J Clin Invest. 2006;116:1974-1982)
If this strategy were to be transferred to human hemophilia gene therapy, HSCs would be harvested after autologous peripheral mobilization and selection, transduced with LV-2bF8, and infused back into the patient. One question would be how much of the hemophilia patient's bone marrow (or HSCs) would need to be treated to have therapeutic efficacy? If this were being done in a patient with a FVIII inhibitory antibody, would the LV-2bF8–transduced HSCs engraft in the presence of preexisting antibody? To answer the first question, 2bF8-transgenic bone marrow was mixed with varying amounts of syngeneic FVIII KO bone marrow to determine the percentage of cells that might need to be transduced to see a therapeutic benefit. Even if only 5% to 10% of the bone marrow contained the transgene, > 60% of the mice survived tail clip challenge.17 Even if the mouse had inhibitory antibody titers as high as 500 to 20,000 BU/mL, bone marrow engrafted and > 75% of the mice survived tail clip.17
Our studies in mice suggest that this approach is feasible for the treatment of human hemophilia subjects, even if they have inhibitory antibodies. Rather than patients with inhibitory antibodies being excluded from gene therapy trails, this approach might target that group specifically.
Platelet-Directed Expression of FIX
Because FVIII could be stored in platelet α-granules without VWF, we wondered if FIX would store in a platelet α-granule and if a megakaryocyte would adequately store γ-carboxylate the synthesized protein. 2b-F9-transgenic mice were developed using a similar strategy to the 2bF8-transgenic mice.18 Two transgenic mouse lines were developed: (1) one with a low level of platelet FIX, 2bF9-L (0.7 mU/108 platelets or 1% of normal whole blood FIX), and (2) a second one with high levels of platelet FIX, 2bF9-H (14 mU/108 platelets or 20% of normal whole blood FIX). Figure 4 demonstrates both lines of normalized survival following tail clip challenge. In contrast to the 2bF8 mice, the 2bF9 mice had small amounts of human F9 in their plasma (2bF9-L, 0.1 U/dL and 2bF9-H, 0.24 U/dL). FIX inhibitory antibodies, unlike FVIII inhibitory antibodies, are known not to require incubation in vitro for titer determination. Not surprisingly therefore, 2bF9 platelet-delivered FIX was not found to be clinically effective in the presence of inhibitory antibodies to FIX (Figure 4, A and D). This suggests that the time dependency of FVIII inhibitory antibodies is critical to its efficacy in the presence of FVIII inhibitors. Thus, platelet FIX may be clinically effective for hemophilia B subjects who do not have inhibitory antibodies, but not for those with inhibitory antibodies. Fortunately FIX inhibitors are not as common as FVIII inhibitors.

Platelet-induced expression of FIX (2bF9) is not as effective as 2bF8 in the inhibitor model. (A) Two transgenic lines of mice were developed that expressed FIX in megakaryocytes and stored FIX in platelet α-granules. The 2bF9 high line (2bF9-H) was equivalent to 20% of normal circulating FIX, and the 2bF9 low line (2bF9-L) corresponded to 1% of normal circulating FIX. Both 2bF9-L and 2bF9-H survived tail-clip challenge similar to wild-type mice. The FIXnull and FIXnull mice infused with 1.6 IU/dL plasma did not survive tail-clip challenge. (B) 2bF9-H mice were subjected to tail-clip challenge in the presence or absence of inhibitory antibody to FIX. Even the 2bF9-H mice had minimal survival when challenged with low or moderate inhibitory antibody. Because these mice have circulating levels at 20% of normal circulating FIX, there is not similar protection from 2bF9 similar to that seen in 2bF8 mice with 1.23% levels of FVIII. Compare results with those for hemophilia A in Figure 3B.
Platelet-induced expression of FIX (2bF9) is not as effective as 2bF8 in the inhibitor model. (A) Two transgenic lines of mice were developed that expressed FIX in megakaryocytes and stored FIX in platelet α-granules. The 2bF9 high line (2bF9-H) was equivalent to 20% of normal circulating FIX, and the 2bF9 low line (2bF9-L) corresponded to 1% of normal circulating FIX. Both 2bF9-L and 2bF9-H survived tail-clip challenge similar to wild-type mice. The FIXnull and FIXnull mice infused with 1.6 IU/dL plasma did not survive tail-clip challenge. (B) 2bF9-H mice were subjected to tail-clip challenge in the presence or absence of inhibitory antibody to FIX. Even the 2bF9-H mice had minimal survival when challenged with low or moderate inhibitory antibody. Because these mice have circulating levels at 20% of normal circulating FIX, there is not similar protection from 2bF9 similar to that seen in 2bF8 mice with 1.23% levels of FVIII. Compare results with those for hemophilia A in Figure 3B.
Endothelial Cells as Targets for Gene Delivery for Hemophilia A
Studies of human endothelial cells have usually utilized umbilical vein endothelium, which has been known for years to synthesize VWF, but not FVIII. Although it has been known that FVIII and VWF associate in plasma and are released together following treatment with desmopressin or 1-deamino-8-d-arginine vasopressin (DDAVP), it was not until recently that human (or murine) microvascular endothelial cells have been demonstrated to synthesize (and store) FVIII.19–21 Previous studies in our laboratory demonstrated that, if FVIII was synthesized in endothelial cells, it was stored together with VWF in Weibel-Palade bodies.22,23 It has been assumed that FVIII was synthesized in the liver and probably within the hepatocyte. Liver, spleen and lymph node transplantation in canine hemophilic dogs was demonstrated to restore FVIII in plasma and reduce clinical bleeding.24–26 Liver transplantation has been used for more than 20 years to treat liver failure and when performed in hemophilia patients, liver transplantation “cures” hemophilia.27 Surprisingly, when hemophilia patients were treated with liver transplantation, FVIII was not released by infusion of DDAVP.28 This suggested that the DDAVP-released pool of FVIII might be from extrahepatic synthesis and stores. By chance, liver transplantation was carried out using a liver donor that had severe hemophilia A. If FVIII was only made in the liver plasma, FVIII would be undetectable; but, in this patient, plasma FVIII levels were normal.29 Taken together, these studies suggest that cells in the liver can make significant amounts of FVIII, but extrahepatic sites exist and the most likely site is the endothelial cell. The relative contribution of hepatic versus endothelial cell synthesis is not known, but this does suggest that the endothelial cell may be an interesting target for gene therapy of hemophilia. Hebbel and coworkers30 demonstrated that human endothelial cells could be grown from circulating endothelial cell precursors in peripheral blood and that these cells could be used for gene therapy of hemophilia. More recently, similar blood outgrowth endothelial cells have been used in canine and murine preclinical studies for hemophilia.31–33
One group has used induced pluripotent stem (iPS) cells using murine tail fibroblasts in the presence of Oct4, Sox2, and Klf4 transcription factors.34 When these induced pluripotent stem cells were subsequently differentiated into endothelial cells, they expressed endothelial-specific markers (CD31, CD34, and Flk1) and began to synthesize and secrete FVIII. When transplanted into the liver of FVIII KO mice, plasma FVIII levels reached from 8% to 12% of normal, and the mice survived the tail-clip bleeding challenge. Besides being a gene therapy approach, this provides further evidence that endothelial cells can synthesize FVIII normally even without transduction.
To study this in greater detail, our group has developed transgenic mice in which the FVIII transgene was placed under the control of the Tie2-promoter. Transgenic mice were bred onto the FVIII KO background, and mice heterozygous for the transgene had levels of FVIII in plasma of 56 U/dL. Mice homozygous for the transgene had plasma FVIII levels of 116 U/dL. These mice exhibited phenotypic correction when challenged with the tail-clip survival test. If these mice were further bred onto the VWF KO mice (ie, Tie2-F8+/– FVIII–/– VWF–/–), plasma FVIII was undetectable. If plasma from a FVIII KO mouse (normal VWF levels) was infused into the Tie2-F8 FVIII–/– VWF–/– mouse, the expected delayed rise in plasma FVIII was seen, and this persisted for > 24 hours. Although plasma FVIII levels were undetectable in the Tie2-F8 double KO mice, these mice all survived the tail-clip challenge. The responses of these mice in the presence of FVIII inhibitory antibodies, however, were strikingly different. In the double KO mice (FVIII–/– VWF–/–), Tie2-F8 was not effective in rescuing the lethal response to the tail-clip challenge.
Expression of FVIIa in Platelets for Hemophilia
Although the primary strategies of the previous approaches is the local delivery of the missing clotting factor, FVIII or FIX, an additional approach has studied the expression of FVIIa in platelets as a possible approach to treat hemophilia patients who have developed inhibitory antibodies.35 Earlier studies demonstrated efficacy of systemic expression of rFVIIa in both murine and canine hemophilia,36,37 but the expression of FVIIa in platelets might circumvent problems with the short half-life of plasma recombinant FVIIa by delivering FVIIa within platelets and might have less chance of systemic thrombogenicity with high plasma levels that might be required with the systemic approach. Interestingly, FVIIa traffics differently from FIX in platelets and targets to the cytoplasm rather than the α-granule. Following platelet activation, the FVIIa translocated to the submembrane and subsequently present on the platelet surface. Using thromboelastography, whole-blood clotting times were shortened and tail-clip mortality reduced even in mice with FVIII inhibitory antibodies. Because there is species specificity for FVIIa and tissue factor interaction, expression of human FVIIa was not effective in the murine model.
Summary
Gene therapy of hemophilia has been studied for many years and has not yet brought the promise clinically of a cure for patients with hemophilia. New approaches to replacement therapy are beginning to use products with enhanced in vivo survival. Early results suggest that such approaches may be very promising. Issues around immunogenicity remain a problem for both replacement therapy, as well as many approaches to gene therapy. Whether the targeting of FVIII to platelets might have clinical efficacy in patients with hemophilia and inhibitors remains unknown. Efficacy and safety in large animal models are still desired by the hemophilia community. Canine models will be the appropriate model for platelet-targeted 2bF8 or similar strategies, but the absence of canine VWF in normal canine platelets may be a barrier to studying efficacy in the face of inhibitory antibodies. A recent new model for hemophilia is the sheep model.38 Because sheep platelets contain ovine VWF, that model may be preferable for conducting preclinical studies for hemophilia gene therapy in the face of inhibitory antibodies. Although new paths to treatment are available, the path to a cure still has barriers that must be overcome. Whether some of these approaches might be applicable to human patients with inhibitory antibodies is not yet known, and additional preclinical studies are still needed.
Acknowledgments
This work was supported by HL-44612 and HL-33721 (RRM), CSL Behring Foundation (RRM) and HL-102035 (QS).
Disclosures
Conflict-of-interest disclosures: The authors declare no competing financial interests.
Off-label drug use: None disclosed.
Correspondence
Robert R. Montgomery, MD, Blood Research Institute, BloodCenter of Wisconsin, Department of Pediatrics, Medical College of Wisconsin, 8727 Watertown Plank Rd., Milwaukee, WI 53226; Phone: (414) 937-3802; Fax: (414) 937-6811; e-mail: bob.montgomer@bcw.edu
