
Abstract
During the last decade, increasing attention has been paid to a unique group of patients with acute lymphoblastic leukemia (ALL) who lie at the crossroad of therapeutic care by pediatric and adult hematologists/oncologists. ALL is a disease that affects infants, children, adolescents, and adult patients. With current therapies, the vast majority of children with ALL are now long-term survivors; unfortunately, the same good results have not yet been obtained for adults with ALL. This review will describe current controversies surrounding the treatment of adolescents and young adults with ALL—a group who finds themselves in the transition from “pediatric” to “adult” treatment approaches. The review focuses on recent insights into disease biology, prognostic factors, and treatment outcomes that have led to a series of prospective clinical trials specifically designed for adolescents and younger adults (AYAs) with ALL. These trials have been designed to provide important new clinical, psychosocial, and biological insights, and to further improve the survival of this challenging and unique group of patients.
Introduction
Acute lymphocytic leukemia (ALL) is a heterogeneous disease, both in terms of its pathology and the populations that it affects. Disease pathogenesis involves a number of deregulated pathways controlling cell proliferation, differentiation, and survival that are important determinants of treatment response.1 Approximately 5200 new cases of ALL are estimated to have occurred in the United States in 2007,2 and survival varies with age and disease biology. Recent data extracted from the Surveillance, Epidemiology and End Results (SEER) registry have demonstrated a statistically significant improvement in survival for older adolescent and adults with ALL (ages 15–59 years) during the past two decades from 1980 to 2004.3,4 The greatest survival improvements occurred in the 15- to 19-year-old age bracket, for whom 5-year relative survival improved from 41.0% to 62.1%, with lower rates of improvement achieved for patients 20 to 59 years of age.3,4 Nevertheless, survival for even the most favorable “young adult” group (the older adolescents) falls short of the outstanding results now achieved in children between the ages of 2 and 10 years, in which nearly 90% are long-term survivors.5 Below, a brief review of the known biological and clinical prognostic features followed by a description of the highly successful treatment of children with ALL lays the groundwork for discussion of the challenge to further improvements in survival for adolescents and young adults (AYAs) with ALL.
Definition of the age range that encompasses the AYA patient is itself controversial, because the age range described in the literature for this population varies depending on the study. This review will largely focus on the group of patients who are 16 to 21 years old at the time of diagnosis, the group of patients most commonly treated by both the pediatric and adult oncologists. Most of the retrospective studies described below have focused on this age group, although the definition of the AYA patient varies, and may include patients up to the age of 30 and, in recent studies, even up to 40 years of age. Prospective studies discussed below that utilize high-risk pediatric ALL regimens are testing this approach in adults up to 60 years of age. These prospective studies will undoubtedly help to define specific biological and therapeutic tolerance and may help us to further refine the optimal treatment for the young adult with ALL.
Biological and Clinical Prognostic Factors in ALL: Setting the Stage for Risk-Adapted Therapy
Of the many variables that influence prognosis, genetic subsets, initial white blood cell count (WBC), age at diagnosis, and early treatment response are the most important.6 Although improved treatment and access to treatment has abolished the prognostic strength of some of the previously described adverse prognostic indicators in pediatric ALL, including male sex, African-American race, and the presence of the t(1;19) E2A-PBX fusion gene, it should be stressed that even so-called low-risk children need a certain degree of treatment intensification to avoid unacceptable rates of relapse.1,7 Table 1 lists the prognostic factors in children and adults that may be used for risk stratification in current clinical trials. In contrast to pediatric ALL, due to the poorer outcomes in adult trials, no group is considered favorable risk; rather, the prognostic groups in adult ALL are distinguished by standard or adverse features.
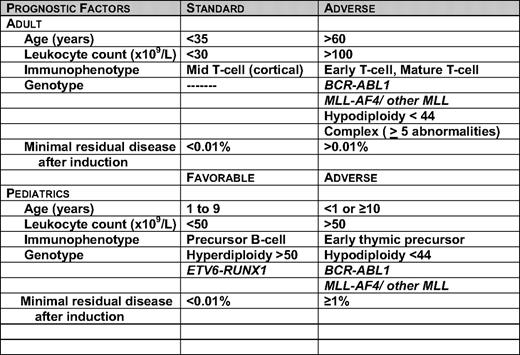
The prognostic impact of age, and to a lesser extent, leukocyte count can be explained by their association with specific genetic abnormalities. For example, there is a preponderance of cases with favorable genetic abnormalities of hyperdiploidy >50 chromosomes or ETV6-RUNX1 in patients 1 to 9 years of age.8 ETV6-RUNX1 mutations are not found in ALL in adults. Adverse genetic abnormalities such as MLL rearrangements occur in 70% to 80% of infant cases, and the Philadelphia chromosome (Ph+) occurs in 25% to 30% percent of adult patients. In a population-based study from the northern United Kingdom of 349 adults diagnosed with ALL over a period of 19 years, Ph+ ALL accounted for 39% of the cases in adults over the age of 60 years.9
Although many genetic abnormalities are associated with clinical outcome, only a few are routinely used for treatment stratification. The Children's Oncology Group also uses trisomy of chromosomes 4, 10, and 17 (triple trisomy) as a favorable prognostic factor in its clinical trials.10 There is clinical heterogeneity within each specific genetic subtype. For example, among patients with Ph+ ALL, patients ages 1 to 9 years fared significantly better than the older patients.11 In adults, Ph+ ALL is associated not only with a high initial leukocyte count, but also with a dismal prognosis with standard chemotherapeutic regimens.12 Allogeneic stem cell transplantation (allo-SCT) in the first complete remission (CR1) has remained a standard curative approach for these adults. In patients with MLL-AF4 fusion, infants and adults have a worse prognosis than children. Approximately 6% of adults with ALL have rearrangements involving the MLL gene.13 The basis of these differences may be related to some combination of secondary genetic events, the developmental stage of the target cell undergoing malignant transformation, and the pharmacogenetics or pharmacokinetic features of the patient. In addition to the t(4;11), the MLL gene is involved in a number of other translocations in ALL, for example, t(11;19) and t(9;11).8 Due to their poor prognosis, allogeneic transplant in CR1 is recommended for adult cases with translocation involving the MLL gene. In one of the largest published series, the outcome of 34 patients with t(4;11) from the LALA-94 study were evaluated.13 Patients with t(4;11) were assigned to allo-SCT in CR1 if a donor was available, with the remaining patients assigned to either autologous stem-cell transplantation (auto-SCT) or intensive post-remission chemotherapy. The outcome for the t(4;11) patients was significantly better for those receiving an allo-SCT in CR1, with a 5 year disease-free survival (DFS) of 66% (±13%) compared with 11% for those receiving an auto-SCT and 20% for those receiving chemotherapy (p = 0.02).
As mentioned above, the significance of many prognostic factors changes with improvements in treatment. For example, the outcome for both children and adults with Ph+ ALL has improved substantially with the addition of tyrosine kinase inhibitors to the treatment.14 Recently, children with this genotype treated with chemotherapy and imatinib (without allo-SCT) had a 3-year event-free survival (EFS) of over 80%.15 If the favorable outcome is confirmed with longer follow-up, the Ph chromosome may join a long list of other factors such as male sex and African-American ethnicity that have lost adverse prognostic impact with improved treatment in childhood ALL.16–18 The recent finding of adverse prognosis of CD20 expression in adult19 but not in childhood ALL20 may have therapeutic implications with the availability of targeted monoclonal antibodies that can be added to front-line therapies.
More recently, the application of microarray-based, genome-wide analysis of gene expression and DNA copy number, complemented by transcriptional profiling, re-sequencing and epigenetic approaches, has identified specific genetic alterations with biologic and therapeutic implications. For example, genetic expression profiling studies have classified T-ALL cases into several distinct genetic subgroups that correspond to specific T-cell development stages: HOX11L2, LYL1 + LMO2; TAL1 + LMO1; or LMO2, HOX11, + MLL-ENL. While HOX11L2 generally confers a poor outcome, HOX11 and MLL-ENL are associated with a favorable outcome.21,22 Among many other mutations in T-cell ALL, NOTCH1 or FBXW7 mutations are associated with a favorable prognosis, and NUP214-ABL1 fusion is responsive to tyrosine kinase inhibition.23–25 Recent genome-wide studies in precursor B-cell ALL identified two high-risk subgroups, one with a genetic profile similar to that of cases with BCR-ABL1 fusion, characterized by IKZF1 deletion, and the other with a JAK2 mutation.26–28 These data suggest potential new therapeutic targets, including gamma secretase inhibitors, to target NOTCH1 mutations and JAK and ABL kinase inhibitors.
Treatment Strategies for ALL
Treatment of ALL involves some of the most complex chemotherapy combinations and treatment schedules used in oncology. Induction chemotherapy is used first to reduce the burden of lymphoblasts in the bone marrow and to restore normal hematopoietic function. Consolidation therapy (also termed intensification) is used with the intention of clearing any drug-resistant leukemia cells that have survived induction therapy and to eliminate minimal residual disease (MRD). Maintenance chemotherapy consists of 2 to 3 years of low-dose antineoplastic drugs designed to prevent leukemia relapse during the crucial few years after remission induction and consolidation. Finally, central nervous system (CNS) prophylaxis is necessary to treat sanctuary sites that are shielded from systemic therapy by the blood-brain barrier. A number of drug combinations and schedules have been empirically developed over the years to bring this multi-step treatment theory into practice. Importantly, the methods by which children and adults are treated have evolved in very different ways.
Cure of ALL in Children: How Did We Get There?
The significant improvements in survival for children with ALL during the past two decades can be attributed to both favorable disease biology and to the success of carefully designed, risk-adapted treatment. Risk stratification evolved during these decades to incorporate response to initial treatment, cytogenetics, and other biological characteristics of the disease. Measurement of MRD, as discussed above, is now used to improve risk stratification and to allocate patients to allo-SCT in many pediatric and in at least one adult clinical trial.29–31
The initial successful strategy was pioneered in the 1980s by the Berlin-Frankfurt-Muenster (BFM) group, who demonstrated that an intensive multi-drug induction and consolidation followed by a delayed intensification phase resulted in improvements in survival to about 70% for children enrolled in these studies.32 The BFM framework consists of: 1) an induction regimen including oral corticosteroids, intravenous vincristine and daunorubicin, intramuscular L-asparaginase, and intrathecal cytarabine and methotrexate; 2) a consolidation regimen of 6-mercaptopurine and intravenous and intrathecal methotrexate; 3) a re-intensification regimen of dexamethasone, vincristine, doxorubicin, and L-asparaginase alternating with cyclophosphamide, cytarabine, 6-thioguanine, and intrathecal methotrexate; and 4) maintenance therapy including oral methotrexate and 6-mercaptopurine for an optimal time of 24 to 36 months.32 Subsequently, as discussed by Smith et al.,5 treatment refinements tested in cooperative group trials identified several key improvements: the addition of an interim maintenance phase, augmentation of the BFM-type delayed intensification, and a reduction in the percentage of children receiving cranial irradiation for CNS prophylaxis.33–35 These strategies, which are focused on extended and intensive use of the “core” drugs glucocorticoids, vincristine, l-asparaginase (and more recently, the use of an extended release PEG-asparaginase,36 methotrexate, and anti-metabolites, have resulted in cure in the vast majority of children with ALL.
Two recent reports from pediatric oncology group trials demonstrate the efficacy of this approach for the AYA patient. In a subset analysis, the Dana Farber Cancer Institute ALL consortium evaluated the treatment outcome for 51 AYA patients ages 15 to 18 years treated on two consecutive trials from 1991 to 2000.37 With a median follow-up of 6.5 years, they reported a 5-year EFS of 78%, which was not statistically significantly different than the outcome of the younger children aged 1–10 years where EFS was 85% (p=.09). The Children's Oncology Group also performed a subset analysis of 262 AYA patients aged 16–21 years enrolled on the CCG 1961 trial from 1996–2002.38 This trial randomly assigned therapies to evaluate the impact of post-induction treatment intensification on outcome. The five-year EFS and OS survival for the AYA patients was 71.5% and 77.5%, respectively. Thus, the outcome for even the highest risk group of patients, the AYAs, is beginning to approach the remarkable cure rates for children with ALL.
Treatment of Adults with ALL During the Past Two Decades
Clinical trials for adults with ALL have been modeled after the trials pioneered by childhood ALL. However, in contrast to the pediatric trials that have been conducted during the past two decades, adult cooperative group trials have generally tested dose intensification of active, myelosuppressive agents such as daunorubicin, cytarabine, and cyclophosphamide and the use of allo-SCT in first remission. The hyper-CVAD regimen designed by the MD Anderson group uses four alternating courses each of cyclophosphamide, vincristine, doxorubicin, and dexamethasone in conjunction with methotrexate/leucovorin and cytarabine for a total of eight cycles of therapy.39 CNS prophylaxis is provided with intrathecal methotrexate and cytarabine. Maintenance therapy with 6-mercaptopurine, methotrexate, vincristine, and prednisone continues for a total of two years.39 The regimen by Larson et al. (CALGB 8811) added cyclophosphamide to the induction regimen of prednisone, vincristine, daunorubicin, and L-asparaginase. In addition, the Cancer and Leukemia Group B (CALGB) investigators added more intensive post-remission therapy that was modeled on pioneering pediatric trials. Their strategy included the use of 6-mercaptopurine, L-asparaginase, and 6-thioguanine along with cyclophosphamide, vincristine, cytarabine, corticosteroids, and methotrexate during early and late intensification after remission. CNS-directed therapy was provided with cranial radiation and intrathecal methotrexate. Maintenance therapy consisted of vincristine, prednisone, oral methotrexate, and 6-mercaptopurine for a total of 24 months after diagnosis.40 In general, the cumulative dose of L-asparaginase and vincristine is lower in adult protocols. All of these adult trials have similar outcomes with overall survival of only 35–40%. These outcomes reflect adults of all ages ranging from 16 years to older than 80 years.
The role of auto-SCT and allo-SCT in first remission was also been explored in a number of trials during this period. A meta-analysis of seven trials that included 1274 patients revealed that there was no beneficial effect of auto-SCT.41 Allogeneic transplant, based on a “donor” vs “no donor” comparison, conferred a survival advantage, particularly for high-risk patients. An evidence-based review panel subsequently recommended allogeneic SCT for ALL in first remission for adults with high risk, but not standard risk disease.42 However, a recently published trial carried out by the Medical Research Council in Great Britain and the Eastern Cooperative Oncology Group (MRC UKALL XII/ECOG 2993 tested the benefit of allogeneic transplant in first remission for adults under the age of 50 years and reported a different outcome than previously published trials.43 In a donor-versus-no donor comparison for the Philadelphia chromosome-negative patients, an overall 5-year survival advantage was noted for patients with a donor of 55% in contrast to 45% for the no-donor group. This survival advantage was noted particularly for the standard risk patients (all under age 35 years with no adverse biologic features) where survival was 62% for those with a donor. In contrast, the high-risk patients (defined as ≥ 35 years old and/or with high risk biologic features), there was no survival advantage for the donor group where treatment related mortality was unacceptable at nearly 36% and overall survival for the donor group was 41% vs 35% for the no donor group. Importantly, this study also demonstrated that auto-SCT in CR1 is not recommended as frontline therapy. Patients receiving auto-SCT in CR1 had an inferior outcome compared to those who received standard consolidation and long-term maintenance chemotherapy.
The AYA Patient: At the Therapeutic Intersection
The trials in both pediatric and adult ALL described above included, but did not specifically focus on the AYA patient. Evaluation of the outcomes of the AYA patient, here defined as patients between the ages of 15–21 years old, presents specific challenges. Because of community referral patterns, these patients may be treated by either pediatric or adult oncologists. As such, the treating physician may view the 15 -21 year old either as an older child or as a younger adult. The oncologist will choose a regimen most appropriate for the population usually seen by that particular physician. A number of comparisons of the clinical outcome of adolescents enrolled on adult and pediatric clinical trials (Table 2) have resulted in interesting observations about what that appropriate treatment regimen should be and have guided the design of a number of prospective clinical trials designed specifically for AYAs with ALL.
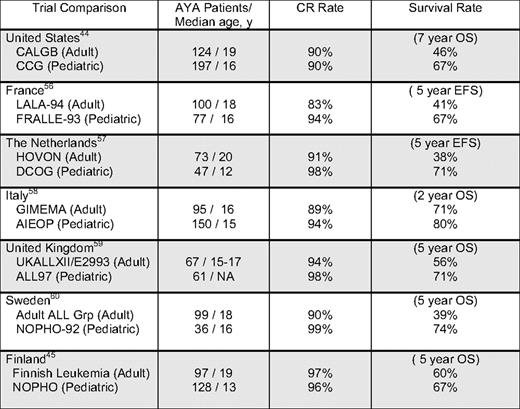
Retrospective Comparison of Pediatric and Adult Cooperative Group Trials in AYAs
The retrospective comparisons summarized in Table 2 were performed by large cooperative groups throughout the world and examined the outcome of the AYA patient treated on pediatric or adult cooperative group trials in ALL that were conducted contemporaneously. The majority of these retrospective comparison studies demonstrated a significant survival advantage for the AYA patient treated by the pediatric vs the adult cooperative group. The first of these trials to have been reported highlights many of the interesting questions posed by these comparisons and will be reviewed briefly below.44 The Cancer and Leukemia Group B (CALGB) and the Children's Cancer Group (CCG) examined the outcome of 321 AYA patients between 16–20 years treated on consecutive trials from 1988 – 2001. The two patient groups were well matched for biological features including immunophenotype and cytogenetics; although the age range was the same in both groups examined, the median age of the patients in the CALGB studies was 19 years compared to 16 years for the CCG patients. CR rates were identical, 90% for both CALGB and CCG AYAs. However, CCG AYAs had a 63% EFS and a 67% overall survival (OS) at 7 years, in contrast to the CALGB AYAs, in which 7-year EFS was only 34% (p < 0.001; relative hazard ratio [RHR] = 2.2]) and the OS was 46% (p < 0.001; RHR = 1.9). Interestingly, the CALGB AYAs 16 to 17 years of age achieved similar outcomes to all CCG AYAs, with 7-year EFS of 55%. In contrast, EFS for 18- to 20-year-old CALGB patients was only 29% (p = 0.01). A difference in pattern of relapse was also noted. The incidence of CNS relapses was significantly higher in CALGB AYAs (11%) compared with the CCG AYAs (1.4%; p < 0.001). While the studies performed by the two groups during this period had similarities in terms of overall treatment plan and schedule, comparison of the planned cumulative doses of drugs administered identified some potentially important differences. CCG patients received considerably more treatment with non-myelosuppressive drugs, including glucocorticoids (both dexamethasone and prednisone), vincristine, and L-asparaginase. CNS prophylaxis was administered earlier, with greater frequency, and for a more prolonged period during CCG treatment. Long-term maintenance therapy was also continued for a longer period in CCG patients.
Since the initial report of these findings in 2000, multiple national European cooperative groups have reported similar results with improvement in the outcome of AYAs treated on pediatric compared with adult protocols; these are summarized in Table 2. Given the retrospective nature of these comparison trials and the significant differences in treatment approach applied by the pediatric and adult treatment groups, it is harder to critically interpret several of these trials. Significant variation in the ages of the patients studied, small numbers of patients in several of these trials, and variations in the regimens utilized, with SCT often applied as a post-remission strategy in the adult European cooperative group trials, make firm conclusions difficult and will require prospective comparisons for validation of these conclusions. Nevertheless, a basic theme emerges from all of these trials: survival was generally significantly better for the young adults treated on pediatric cooperative group trials. Similar to the CALGB and CCG comparison, the major treatment differences included significantly greater cumulative dosing of the glucocorticoids, vincristine, and l-asparaginase, and more intensive CNS prophylaxis.
The results of a retrospective comparison of patients treated in Finland differ from those from the studies described above. In this study,45 a group of 128 patients from 10 to 16 years of age (median age, 12.9 years) treated on pediatric protocols were compared with 97 patients 17 to 25 years of age (median age, 18.9 years). Despite the significant difference in median age and some differences in drug schedule and dosing between the two groups, there was no significant difference in the 5-year EFS (67% for the pediatric group and 60% for the adult group) or OS (77% in the pediatric group and 70% for the adult group). Interestingly, in this comparison, there were no significant differences in the doses of corticosteroids, vincristine, or asparaginase between the pediatric and adult trials, although the pediatric trials contained higher cumulative doses of methotrexate and, in the adult protocols, more anthracycline. Similarly, retrospective data from the MD Anderson Cancer Center using the HYPER-CVAD regimen have also reported favorable results in their AYA patients. With a median age of 19 years, the CR rate was 97% for 102 AYAs with newly diagnosed ALL and OS was 65%.46 In both the Finnish study and the MD Anderson study, all patients were treated in large referral centers for treatment of leukemia. One possible interpretation of these retrospective analyses is that treatment outcomes may be better regardless of nuances in chemotherapy dose and schedule when patients are treated at larger centers with greater experience and expertise in the administration of these complex regimens.
Hints to Differences in Outcome for AYAs: Treatment/Pharmacodynamics/Compliance?
As alluded to above, several potential explanations exist for the striking differences seen in the majority, but not all, of the retrospective comparisons. In addition to clear differences in protocol design and dose intensity, highlighted above, other explanations include interesting new pharmacodynamic insights, clinical and demographic differences in AYAs receiving treatment at pediatric compared with adult centers, and potential variations in the degree of adherence to protocol drug administration by adult compared with pediatric oncologists.
As highlighted in the CALGB versus CCG comparison, while the adult trials often focused on dose intensification of the myelosuppressive agents, including cyclophosphamide and anthracyclines, the pediatric regimens employed significantly higher cumulative doses of the drugs that have made up the backbone of the BFM model: glucocorticoids, vincristine, and l-asparaginase. CNS prophylaxis in the pediatric regimens always begins during induction therapy and continues during maintenance therapy, achieving a higher cumulative dose than the majority of the adult trials. Overall, the pediatric regimens also continue long-term maintenance for a longer period, particularly for male patients.
Recent studies are beginning to shed new light on potential biological differences in AYA patients. Metabolism of some of the key drugs may begin to change during late adolescence, resulting in potential increases in drug-related toxicities and/or decreased exposure to critical drugs such a l-asparaginase and dexamethasone.47 There are few studies quantifying the tolerability of l-asparaginase in young adults. In a preliminary review of the serious toxicities reported in the first 112 patients enrolled in the new North-American intergroup study C-10403, which includes patients 16 to 39 years of age, there were serious adverse events attributed to PEG-asparaginase, including hypersensitivity (11%), coagulopathy (20%), and pancreatitis (3%) (W. Stock and K Donohue, personal communication). Data from children treated on the Dana Farber 91–01 trial showed that older children (9–18 years) had a significantly higher incidence of thrombosis compared with younger children (15% vs. 2%; p < 0.01).7 Thus, the ability to actually deliver the dose intensity of the agents that contribute to successful outcomes may be more challenging as patients enter their young adult years.
The degree of myelosuppression achieved during long-term maintenance has also been demonstrated to be of particular importance to EFS for adolescents with B-lineage intermediate risk ALL.48 While nearly all collaborative group pediatric ALL protocols recommend adjustment of the doses of methotrexate and 6 mercaptopurine during maintenance therapy to be adjusted to a target WBC level, few adult cooperative group trials have recommended this dose adjustment.
One of the more intriguing issues posed by these retrospective comparisons is the potential contribution of disparity in the practice patterns of the pediatric and adult hematologists/oncologists and patient compliance to the outcome of AYA patients. This topic has been discussed heatedly at recent hematology meetings and in the literature,49 yet the question remains largely unanswered. Virtually all children with ALL are referred to pediatric centers and treated in clinical trials by pediatric oncologists who are focused on the treatment of ALL and have highly skilled support staff who carefully monitor protocol compliance. In contrast, ALL is a rare disease for adult hematologists/oncologists, for whom familiarity and compliance with the complex ALL treatment regimens are likely to pose a significant challenge. Of note, in the Finnish trial where no significant differences in outcome were noted between pediatric and adult trials, all patients were treated at one of five central referral hospitals in Finland, possibly ensuring a higher degree of familiarity and compliance to treatment. Finally, due to differences in referral patterns to pediatric or adult centers, most of the comparison trials show a significant skewing of age, with a significantly lower median age in the pediatric AYA patient compared with the adult AYA patient. From the CALGB versus CCG comparison, in which the youngest CALGB AYAs (16–17 years of age) fared as well as the CCG AYAs compared with the 18- to 20-year-old CALGB AYAs, it is intriguing to postulate that the significantly worse outcomes in only the older CALGB AYAs could result from the that that the 18- to 20-year-olds are living independently and removed from family members who might ensure greater compliance with protocol medications and outpatient follow-up. Others have suggested that the older AYAs may face significantly more challenges in access to health care due to insurance issues and prescription drug coverage for the crucial outpatient medications that are the mainstays of therapy.50
Prospective Trials: Moving Forward
To begin to address the many unanswered questions that have been raised by the retrospective comparison trials and to determine whether AYA patients treated by adult hematologists/oncologists can achieve similarly improved outcomes to the pediatricians for this age group, prospective cooperative group trials in North America and in Europe have begun, and early results from several of these trials have recently been reported. The Program Español de Tratamiento en Hematología (PETHEMA) Protocol ALL-96 addressed the toxicity and results of a pediatric-based protocol in 35 adolescent (age 15–18 years) and 46 young adults (age 19–30 years) with standard-risk ALL.51 In this trial, patients received a standard five-drug, five-week induction course, followed by two cycles of early consolidation, maintenance with monthly reinforcement cycles for one year following remission, and standard maintenance chemotherapy for up to two years following CR. The adolescents and young adults were well-matched for pre-treatment characteristics. The CR rate was 98% and with median follow-up of 4.2 years, the 6-year EFS and OS were 61% and 69%, respectively. The only significant predictor of poor EFS for the entire group was a slow response to initial induction therapy (>10% blasts remaining in bone marrow aspirate done on day 14 of induction therapy). Thus, the investigators concluded that a pediatric regimen was tolerable and efficacious in AYAs with ALL up to the age of 30 years.
Two pilot studies from French adult cooperative groups have also demonstrated the feasibility of employing modified pediatric-inspired regimens in adults with ALL, although the age range of these studies extended well into the middle years of adult life. In the FRALLE study, 28 Philadelphia chromosome-negative adult ALL patients 16 to 57 years of age were treated on the FRALLE 2000 protocol, which consisted of a prednisone prophase and a four-drug induction including L-asparaginase, consolidation, delayed intensification, and maintenance chemotherapy. The 4-year DFS was 90% versus 47% seen in matched historical controls.52 In the GRAALL-2003 study, 225 patients with Philadelphia chromosome-negative ALL 15 to 60 years old (median, 31 years) were treated between 2003 and 2005 with five drug-induction, dose-intense consolidation, delayed intensification, and two-year maintenance therapy.53 Notably, allo-SCT for patients ≤ 55 years was recommended in this trial and makes interpretation of this trial more problematic. The CR rate was 93.5%. Among the 139 CR patients, 71 actually underwent transplantation in CR1 and were censored at the time of transplant. At 42 months, EFS was 55% versus 41% when comparing patients from an earlier French trial, the LALA-94, and OS was 61%, significantly better than 41% OS in the LALA-94 (p<0.001). The benefit of the GRAALL approach was not statistically significant in patients older than 45 years due to a significant increase in treatment-related mortality of 23% in comparison to 5% TRM for patients < 45 years old. The investigators concluded that the use of a pediatric inspired regimen in adults up to 45 years old was tolerable and markedly improved outcome for “younger” adults with ALL. As mentioned above, several factors must be considered when interpreting these results – this regimen included older adults up to the age of 60 years and utilized only a modified pediatric regimen that did not employ the dose intensity of corticosteroids, asparaginase and vincristine that are routinely used in current pediatric regimens. Another difference from the pediatric regimens is that all patients on these trials still received prophylactic cranial irradiation. Also, the majority of CR1 patients actually underwent allo-SCT, which is not the approach employed by pediatric groups. Thus, any interpretation of the contribution of the “chemotherapy” intensification component of these trials to DFS is very difficult.
In contrast, the Dana-Farber Cancer Institute consortium has also extended their successful pediatric regimen37 to older patients 18–50 years old. The trial design here was a true pediatric approach with intensification of E coli l-asparaginase, glucocorticoids and vincristine for patients up to 50 years old – allo-SCT was not routinely recommended. Early results from this completed phase II trial were presented at the 2007 ASH meeting and long-term follow-up is now available (Dr. Dan DeAngelo, maunuscript in preparation 2010, and personal communication).54 Ninety-four patients were evaluable, with a median age of 28 years (range 18–50). Seventy-nine patients (84%) achieved a CR after one month of intensive induction therapy. With a median follow-up time of 45 months, their group reports an estimated DFS rate for all patients of 66% and OS rate of 65%. For the 74 patients with Philadelphia chromosome-negative ALL, DFS was 70% and OS was 68%.
The largest prospective trial to evaluate the feasibility of utilizing a pediatric regimen in AYA patients treated by adult medical hematologists/oncologists is ongoing in North America (C-10403). The US adult and pediatric cooperative groups have enrolled over 125 of a planned 300 patients on a prospective phase II trial for AYAs from 16–39 years old (C-10403) that utilizes one treatment arm of the current COG study (AALL0232) for adolescents (and high-risk children with ALL).35,38 This study will examine prospectively molecular genetics, MRD, psychosocial disparities and treatment adherence on the part of physicians and patients referred to and treated on an intensive pediatric regimen by adult oncologists. Importantly, the ability to administer in a safe and timely manner the same treatment that is being used by the US pediatric cooperative group will be examined. Treatment toxicities in addition to schedule and dose compliance are being collected prospectively. Each patient participates in a baseline and follow-up questionnaire to explore psycho-social and socio-economic conditions to begin to gather critical information about the other critical issues that likely influence treatment outcomes.
These prospective trials are demonstrating that it is possible to achieve significant improvements in outcome for AYAs with ALL although many challenges remain to ensure access to care, to minimize treatment toxicity and to develop and follow specific survivorship monitoring plans. While fewer specific data are available on long term complications of survivors of young adults with ALL, the long term complications of successful treatment of children with ALL have been well described and include neurocognitive and neurologic dysfunction, endocrine and metabolic abnormalities (including obesity), bone toxicity (osteonecrosis), cardiac toxicity and secondary malignancies.55 To guide the frequency and focus of medical visits, and the ordering of appropriate surveillance tests, comprehensive guidelines have been published in many countries, including North America, where guidelines created by the Children's Oncology Group, entitled “Long-Term Follow-up Guidelines for Survivors of Childhood, Adolescent and Young Adult Cancers” are available at www.survivorshipguidelines.org and should be adopted by the adult oncology community. The next generation of studies, linked to important biological, psychosocial and pharmacological correlates, will result in further insights into optimizing the comprehensive approach to treatment and follow-up for this significant group of patients with ALL and may be the next step to achieving the high cure rate and successful transition back to “normal” life now routinely achieved in children with ALL.
How I Treat
A 20 year old college student is admitted to the adult oncology ward for treatment of newly diagnosed ALL. Initial CBC showed a WBC of 2.3 K/μl, hemoglobin of 9.4 gm/dL, and platelet count of 19K/μl. A bone marrow examination demonstrates precursor B-lymphoblastic leukemia extensively involving a hypercellular marrow. Flow cytometry had demonstrated that the blasts were CD19+, CD20+, CD10+, CD79a+, CD51+. CD34+ and Tdt+. Cytogenetics revealed a normal male karyotype. LDH is 850 u/L.
This young adult with ALL has presented with standard risk features. Based on the preponderance of data presented in the review, the best therapeutic approach for this patient would be an intensive pediatric regimen with intensive glucocorticoids, vincristine, and PEG-asparaginase with early and frequent CNS prophylaxis. Prolonged maintenance therapy with adequate myelosuppression is also a key feature of a pediatric regimen. While the ECOG/MRC data have suggested that a young adult patient has improved survival with allo-SCT compared to their standard chemotherapy regimen (which was not a pediatric-like regimen), there are no data that have unequivocally shown the benefit of an allogeneic transplant for this patient. Thus, I would ask this patient to participate in the current intergroup phase II trial, C-10403 (www.clinicaltrials.gov) that is a phase II trial for adolescents and young adults with ALL that is being conducted in collaboration with the Children's Oncology Group Study AAALL0232 and which utilizes a regimen that has resulted in an EFS of 72% in 262 patients 16–21 years old.
The patient consents for the trial. I would give induction therapy as an in-patient since sudden sepsis and early treatment toxicities from the multi-drug induction therapy are common. This patient should be treated with allopurinol for 10 days to prevent hyperuricemia and complications of tumor lysis. Rasburicase administered prior to initiation of chemotherapy should also be considered. Routine anti-viral and PCP prophylaxis are administered throughout the treatment. It is important to remember that no sulfa drug (or non-steroidals, etc) should be given on days that he receives methotrexate since there many commonly used drugs that can interfere with renal excretion of methotrexate. It is mandated in the protocol that all patients are pre-medicated with hydrocortisone and tylenol prior to each dose of PEG-asparaginase to try to lessen the risk of hypersensitivity reactions. No routine anti-thrombotic prophylaxis is recommended; there are some data from pediatric groups suggesting that the use of anti-thrombin III treatment may be useful but this has not been routinely employed by the adult oncology community and there are no prospective studies. Cryoprecipitate should be administered for fibrinogen levels < 50 to avoid hemorrhagic complications. The patient has an uncomplicated induction course and has a rapid early response to treatment documented by the day 15 of induction therapy bone marrow examination. Other than some mild agitation and hyperglycemia which are medically managed, his course is uncomplicated and he achieves a CR on d 28 of induction therapy which is documented by bone marrow examination and flow cytometry for MRD.
Following recovery from the induction therapy, the remainder of therapy is administered as an outpatient. We routinely create a detailed calendar for the patient. Together, nursing staff and physicians routinely review supportive care meds, encourage patients to keep a treatment diary, and always try to have the same caregivers in our IV therapy suite to establish and maintain continuity and close rapport. As all patients and caregivers know, the successful treatment of ALL depends heavily on commitment to medication compliance and protocol adherence – and these are often tall orders to fill, particularly for young adults who are seeking independence and freedom from constant supervision. Thus, it truly “takes a village” to achieve successful completion of these arduous, but highly successful protocols. Frequent laboratory and physical examinations are important to assess for the toxicities of the treatment particularly the hyperglycemia, liver dysfunction, and peripheral neuropathy that can be cumulative during these prolonged therapies. Our social worker and staff psychologist are key caregivers for these young adults and meet with the patient routinely during his follow-up visits. Routine yearly bone density tests are monitored. As this patient continues during long-term remission, we would advise following survivorship monitoring guidelines as recommended by the pediatricians and referenced above in the text.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interest.
Off-label drug use: Alemtuzumab and rituxan may be discussed in clinical trials of ALL.
Correspondence
Wendy Stock, MD, University of Chicago Medical Center, 5841 S. Maryland Ave., MC 2115, Chicago, IL 60637; Phone: (773) 834-8982; Fax: (773) 702-0963; e-mail: wstock@medicine.bsd.uchicago.edu
