
Abstract
Systemic light-chain amyloidosis (AL) is caused by misfolded immunoglobulin light-chain proteins that aggregate and deposit as unique fibrils, ultimately leading to organ failure and death. Recent developments that have significantly aided the management of patients with AL include diagnostic techniques for definitive typing of amyloid deposits by use of laser microdissection with mass spectrometry and customized protein bioinformatics, and validated staging and response-scoring systems that improve clinical trial design. The widespread use of cardiac biomarker staging and serum-free light-chain levels to evaluate response to therapy has also improved care. Standard therapies such as oral melphalan and dexamethasone or autologous stem cell transplant continue to be important options, while thalidomide and its analogs, lenalidomide and pomalidomide, and the proteasome-inhibitor bortezomib have activity in AL and have expanded our armamentarium. Continued improvement in outcomes, however, will require the commitment and cooperation of pharmaceutical companies, regulatory agencies, academic investigators, and cooperative groups/consortia. This effort will involve the conduct of well-designed clinical trials of new agents and combinations within a modern framework that categorizes the study populations of patients with AL, defines the end points appropriate to those populations and to the different phases of clinical trials, employs the newly available staging and response criteria, and standardizes adverse event reporting.
Introduction: A Proteotoxic Clonal Plasma Cell Disease
The systemic amyloidoses share a common pathophysiologic mechanism: systemic proteotoxicity caused by aberrant precursor molecules whose misfolded intermediate forms are often toxic to cells and whose aggregates deposit as interstitial fibrils of amyloid.1 These processes lead to organ dysfunction and death, most notably from cardiac involvement. The most common type of systemic amyloidosis is immunoglobulin light-chain amyloidosis (AL), with an incidence of 8 to 10 cases per million person-years, a median age at diagnosis of 63, and a median survival time if left untreated of 12 months.
The next most common type of systemic amyloidosis are the transthyretin (ATTR) types caused by either mutant (hereditary) variants or wild-type (“senile systemic”) transthyretin. Secondary amyloidosis, although rare in developed countries, still occurs with autoimmune or inflammatory diseases, malignancies (e.g., renal cell cancer), and chronic infections. Other hereditary types have also been described and may confound the diagnosis of AL. In this review, we focus on AL, discussing recent advances, the results of recent clinical research, and the need for a framework to plan pivotal trials for new therapies. AL remains an “orphan disease” whose patients comprise a half-dozen potential study populations with unmet pharmacologic needs. At a time when the road to approval for new agents for myeloma has become crowded and more challenging, it is sobering to note that there are no drugs approved for AL. Even oral melphalan can be difficult to obtain without claiming that the patient has myeloma.
Making the Diagnosis and Getting the Type Right
Because clinical presentations are similar for the different types of amyloidosis, the stigmata of organ involvement do not enable one to deduce the type in any given patient (Table 1). Two potential precursor proteins can coexist in a patient, making it perplexing as to which type of amyloidosis is present; this is particularly relevant in the African-American population, which has higher rates of monoclonal gammopathies and a 4% incidence of an hereditary ATTR variant (Val122Ile), and in elderly men, who also have higher rates of both monoclonal gammopathies and wild-type ATTR.2,3 In these cases, patients almost always have one type of amyloid causing the disease, and confluent risks exist for both doctor and patient: failing to diagnose a hereditary disease that also affects the patient's family; treating with chemotherapy inappropriately; or not treating in a timely way or at all. Fortunately, recent advances in diagnostic testing can identify the type of amyloidosis with high reliability and accuracy.
Types of systemic amyloidosis* and patterns of organ involvement by type
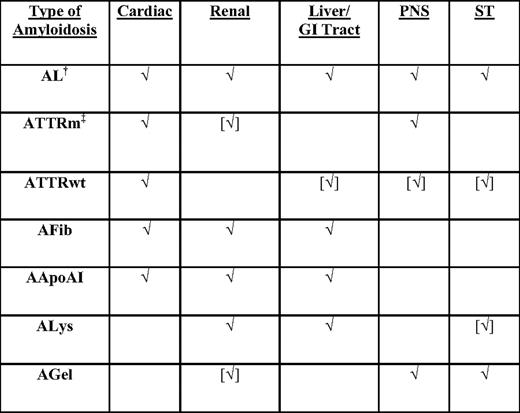
√, organ commonly involved; [√], organ uncommonly involved; AApoAI, mutant apoliprotein A-I amyloidosis; AFib, mutant fibrinogen A-α amyloidosis; AGel, mutant gelsolin amyloidosis; ALys, mutant lysozyme amyloidosis; ATTRm, mutant transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis; PNS, peripheral nervous system; ST, soft tissues.
*The current nomenclature of amyloidosis is based on the nature of the major fibrillar protein that is designated protein A, followed by an abbreviation of the protein's name. The protein must be a major fibril protein in extracellular deposits with affinity for Congo red with apple-green birefringence.
†Monoclonal gammopathies are two times more common in African Americans than in Caucasians.
‡V122I ATTRm occurs in 4% of African Americans.
Amyloidosis remains a tissue diagnosis. Tissue biopsy, either of an involved organ or a surrogate site (e.g., abdominal fat), must demonstrate amyloid deposition by classic Congo red staining or electron microscopy. For typing, immunohistochemical staining is frequently unreliable and inaccurate, and immunogold electron microscopy (IEM) is reliable but limited by serologic dependence.2 Laser microdissection with mass spectrometry (LMD/MS) with customized bioinformatic assessment of the constituents of the Congophilic deposits is now the gold standard for typing amyloid, enabling precise identification of type in over 98% of cases (Figure 1).4 LMD/MS is specifically indicated for typing in cases in which two potential amyloid precursor proteins are present in a patient. A critical feature of LMD/MS is that it captures all of the chaperone and fellow-traveler elements in amyloid deposits, as well as the identity of the protein in the fibrils. LMD/MS therefore possesses an internal validation unavailable for any other tissue- or protein-based technique. LMD/MS data, in the aggregate and correlated with clinical and outcome data, may lead to a significant increase in knowledge based on discovering the proportions and polymorphisms of the constituents of amyloid deposits. DNA sequencing of genes related to hereditary variants is also useful, particularly for confirming proteomic findings and subsequent screening of kin.3 The best route to diagnosis, however, is still a physician's thinking of the diagnosis and obtaining or, in the case of the pathologist, appropriately staining, the tissue biopsy.

MS-based proteomic classification of amyloidosis. Amyloid deposits were identified on Congo-red-stained paraffin sections under fluorescent light and microdissected with a laser. The microdissected amyloid fragments were then digested into tryptic peptides, separated by nano-flow liquid chromatography, ionized by electrospray ionization, and analyzed by tandem MS/MS. MS raw data files were queried by the use of three different algorithms (Mascot, Sequest, and X!Tandem), and the results were assigned peptide and protein probability scores. The proteins are listed according to the abundance with which they were represented in the sample based on spectral counting. The amyloid-forming protein identified is immunoglobulin lambda light chain (red boxes; Ig lambda chain constant and V-I regions), which is consistent with AL (lambda-type) amyloidosis. In addition, the samples contain several proteins known to be associated with amyloid deposits, such as apolipoprotein E and the serum amyloid P component, as well as stromal proteins such as actin, vitronectin, and vimentin. The identity and amounts of chaperone and fellow-traveler proteins cannot be precisely determined by any other technique and, once data are available on a large number of annotated cases, correlative analyses will likely reveal patterns of disease and progression linked to aspects of these accompanying proteins, such as proportions and polymorphisms. (Images courtesy of Dr. Ahmet Dogan, Mayo Clinic, Rochester, MN; dogan.ahmet@mayo.edu. See Vrana et al., 20094 for more information.)
MS-based proteomic classification of amyloidosis. Amyloid deposits were identified on Congo-red-stained paraffin sections under fluorescent light and microdissected with a laser. The microdissected amyloid fragments were then digested into tryptic peptides, separated by nano-flow liquid chromatography, ionized by electrospray ionization, and analyzed by tandem MS/MS. MS raw data files were queried by the use of three different algorithms (Mascot, Sequest, and X!Tandem), and the results were assigned peptide and protein probability scores. The proteins are listed according to the abundance with which they were represented in the sample based on spectral counting. The amyloid-forming protein identified is immunoglobulin lambda light chain (red boxes; Ig lambda chain constant and V-I regions), which is consistent with AL (lambda-type) amyloidosis. In addition, the samples contain several proteins known to be associated with amyloid deposits, such as apolipoprotein E and the serum amyloid P component, as well as stromal proteins such as actin, vitronectin, and vimentin. The identity and amounts of chaperone and fellow-traveler proteins cannot be precisely determined by any other technique and, once data are available on a large number of annotated cases, correlative analyses will likely reveal patterns of disease and progression linked to aspects of these accompanying proteins, such as proportions and polymorphisms. (Images courtesy of Dr. Ahmet Dogan, Mayo Clinic, Rochester, MN; dogan.ahmet@mayo.edu. See Vrana et al., 20094 for more information.)
Staging, Prognosis, and Responses to Treatment in Systemic AL Amyloidosis
Once amyloidosis has been demonstrated by biopsy, subsequent work-up involves the identification of the precursor protein and determination of the extent of organ involvement. Testing for a monoclonal gammopathy is performed with serum and urine protein studies and bone marrow biopsy, with immunohistochemical staining for CD138, kappa, and lambda, and Congo red staining for amyloid. The serum-free light-chain assay is critical for evaluating and monitoring patients with AL, because many patients lack a circulating intact immunoglobulin, and serum and urine immunofixation have limited sensitivities.5 The use of all three assays can identify the paraprotein in 99% of patients.6 Rarely, the amyloid-forming light chain is produced by a low-grade lymphoproliferative disorder, and therapy then should be tailored to treat the underlying B-cell disorder.7 If this work-up fails to identify a clonal B-cell disorder, then the AL type is highly unlikely and evaluation for other types should commence. If the work-up reveals a monoclonal gammopathy, the type is almost always AL, although more data should be obtained with IEM, LMD/MS, or TTR gene sequencing (TTRgs) in African-Americans (TTRgs), elderly men (IEM or LMD/MS), those with dominant peripheral or autonomic neuropathy (TTRgs; a common presentation of hereditary types), patients with a family history of amyloidosis (TTRgs), and those with coexisting inflammatory disorders (IEM or LMD/MS).
Staging for cardiac involvement is a critical part of the initial evaluation of amyloidosis. Criteria for the assessment of organ involvement at baseline and of organ response after treatment have been standardized.5 Prognosis in systemic AL amyloidosis remains a function of the extent of cardiac involvement, with a median survival of 6 months for untreated or non-responding patients with symptomatic cardiac AL.8,9 Patients with cardiac involvement can present with fatigue, progressive dyspnea on exertion, findings of diastolic dysfunction, left ventricular hypertrophy in the absence of hypertension, and low voltage on electrocardiogram.5 Serum troponins (I or T) and B-type natriuretic peptides (either BNP or NT-proBNP) are highly sensitive markers of cardiac involvement, and normal values exclude clinically significant cardiac amyloid.10 Cardiac MRI is emerging as a useful tool, particularly in patients with left ventricular hypertrophy and prior hypertension or valvular heart disease, although large studies defining its role are lacking at this time.11 The cardiac biomarkers NT-proBNP and troponin T (or I) are prognostic with respect to survival in AL patients.10,12 A cardiac risk assessment or “cardiac-staging” system incorporating these biomarkers is currently in use, with patients assigned to stage I, II, or III based on the presence of 0, 1, or 2 of the biomarkers, respectively, exceeding threshold levels (NTpro-BNP > 332 ng/L; troponin T > 0.035 μg/L). Patients in these three stages differ significantly with respect to survival (Figure 2A).12 At this time, biomarker criteria for clinical cardiac improvement and progression after therapy are being incorporated into the consensus criteria for organ response.13 Post-therapy, in patients with cardiac involvement, a greater than 30% reduction and a greater than 300 ng/L decrease in the NT-proBNP level from baseline are correlated with improved overall survival, while increases of that magnitude are correlated with progression and worse survival (Figures 2B, C).13

Cardiac and hematologic responses and survival in AL patients. (A) Survival according to “cardiac stage” in 242 patients with cardiac AL. Thresholds for cTnT and NT-proBNP are > 0.035 μg/L and > 332 ng/L. (Reprinted with permission from Dispenzieri et al., 200412 © 2008 American Society of Clinical Oncology. All rights reserved.) (B, C) NT-proBNP response and progression criteria. Annotated data on 685 AL patients from six countries were pooled and analyzed in order to define and validate cardiac biomarker criteria.14 (D) Updated event-free survival in the landmark analysis of the phase III MDex vs. SCT trial.24 Eleven patients in the MDex arm (eight in continuous CR) and five patients in the SCT arm (three in continuous CR) have yet to receive second-line therapy. Initially there were 50 patients in each arm. (Courtesy of Dr. Arnaud Jaccard and co-investigators.) (E) Hematologic response criteria and survival. Patients in the international case series were analyzed for survival after therapy based on modified metrics for hematologic response13 ; 43.6% received MDex, 11.4% SCT, 22% IMiD-based therapy and only 3% bortezomib and dexamethasone. Response criteria were: CR (immunofixation negative and FLC normal), VGPR (very good partial; reduction in the dFLC to < 40 mg/L), PR (partial; reduction in the dFLC by > 50%), and NR (no response; less than PR). (Courtesy of Dr. Giovanni Palladini and co-investigators.)
Cardiac and hematologic responses and survival in AL patients. (A) Survival according to “cardiac stage” in 242 patients with cardiac AL. Thresholds for cTnT and NT-proBNP are > 0.035 μg/L and > 332 ng/L. (Reprinted with permission from Dispenzieri et al., 200412 © 2008 American Society of Clinical Oncology. All rights reserved.) (B, C) NT-proBNP response and progression criteria. Annotated data on 685 AL patients from six countries were pooled and analyzed in order to define and validate cardiac biomarker criteria.14 (D) Updated event-free survival in the landmark analysis of the phase III MDex vs. SCT trial.24 Eleven patients in the MDex arm (eight in continuous CR) and five patients in the SCT arm (three in continuous CR) have yet to receive second-line therapy. Initially there were 50 patients in each arm. (Courtesy of Dr. Arnaud Jaccard and co-investigators.) (E) Hematologic response criteria and survival. Patients in the international case series were analyzed for survival after therapy based on modified metrics for hematologic response13 ; 43.6% received MDex, 11.4% SCT, 22% IMiD-based therapy and only 3% bortezomib and dexamethasone. Response criteria were: CR (immunofixation negative and FLC normal), VGPR (very good partial; reduction in the dFLC to < 40 mg/L), PR (partial; reduction in the dFLC by > 50%), and NR (no response; less than PR). (Courtesy of Dr. Giovanni Palladini and co-investigators.)
In addition to cardiac involvement, the other important prognostic factor for amyloidosis is the response to therapy. Higher baseline absolute values of the involved free light chain (iFLC), as measured by the serum-free light-chain assay, are associated with a greater degree of organ involvement and worse overall survival.14 Post-therapy, achievement of a hematologic response remains the critical variable for predicting organ improvement and for prolonged survival.15 Decreases in the iFLC with therapy are strongly associated with improved survival. Serial iFLC measurements with each cycle of therapy provide the best assessment of therapeutic efficacy or lack thereof.16–18 The goal of therapy is typically a complete hematologic response (CR). Often we aim for CR (characterized by undetectable M protein, normal marrow, and normalization of iFLC levels and the κ-to-λ ratio) with, for example, adjuvant therapy in patients who have persistent monoclonal disease after stem cell transplant.7,19 In patients with significant comorbidities or treatment-related side effects, a 90% reduction in iFLC is likely a reasonable target. Failure to achieve a 50% reduction in the iFLC has been associated with significantly shortened survival,16 so one could argue for changing or intensifying therapy in patients with multi-organ involvement who fail to achieve at least a 50% reduction in iFLC levels after several cycles of therapy. While achievement of a complete hematologic response remains the ideal, the achievement of both hematologic and organ responses or stabilization of organ disease remains a clinically acceptable outcome in many cases. Modified criteria for scoring hematologic response based in part on reductions in the difference between involved and uninvolved FLC (dFLC) have been validated in the large international case series that is the basis for consensus criteria emerging from the recent XII International Symposium on Amyloidosis (Figure 2E).13
It is important to note that 10% to 15% of AL patients have minimally abnormal FLC with either small, absolute numerical dFLC or elevated FLC with normal ratios. In these cases, a response-scoring algorithm based solely on FLC is not useful for clinical care and is too restrictive for clinical research. Patients whose hematologic response cannot be scored by FLC changes, such as those with renal insufficiency, remain evaluable based on traditional M-protein and marrow criteria, in some cases for M-protein responses and in others for CR only (normalization of the FLC ratio and/or of serum and urine immunofixation and marrows). Similarly, patients with small M proteins (e.g., ≤0.2g/dL) and normal FLC may only be evaluable for CR. Despite these complexities, objective measures of organ response and progression will allow reliable clinical assessments and guide therapy over time. From the perspective of clinical research, progression-free survival (PFS) and overall survival (OS) remain key measures of outcome as well.
Clonal AL plasma cells harbor recurring cytogenetic abnormalities, including t(11;14), gain 11q, del 13q, and gain 1q, while t(4;14) and del 17p are not typically seen.20,21 Interestingly, t(11:14) was associated with worse overall survival in a recent study of AL patients,21 in contrast to its association with a relatively favorable prognosis in myeloma. Moreover, we have recently demonstrated that overexpression of cyclin D1 (CCND1 located on chromosome 11q13) in purified AL plasma cells occurs in one-half of AL patients and is associated with unique pathobiologic characteristics at diagnosis: high frequencies of light-chain-only M proteins (not intact Ig M proteins) and kappa light chains, increased cardiac biomarker levels, and poorer overall survival.22 Further validation of these findings, and future incorporation of such new prognostic factors into staging systems, will allow for risk stratification and eventual refinement of therapeutic options based on objective, disease-related variables.
Frontline Therapy: The Newly Diagnosed Patient
Therapies for AL are aimed at eliminating the clonal plasma cells producing the toxic precursor protein. With current approaches, organ improvement may occur in those who achieve at least a partial hematologic response, with kidney and liver responses occurring most commonly. In one series, achievement of a more than 90% reduction in the iFLC or of a CR was associated with organ improvement over 90% of the time.18 Organ responses can lag 6 to 12 months behind the hematologic response (reduction of the iFLC), necessitating aggressive supportive care and collaborative management with other subspecialists, particularly in patients with advanced cardiac or renal involvement.
Oral melphalan and dexamethasone (MDex) is a standard frontline therapy, inducing a hematologic response 67% of the time, with 33% CR and an organ-response rate of 48% in a phase II study of 45 patients. An update of this cohort, a 5-year follow-up, showed an impressive median PFS of 3.8 and OS of 5.1 years.23 Subsequent studies have confirmed the efficacy of this regimen,24 although outcomes for symptomatic cardiac AL patients remain relatively poor (median OS, 10.5 months).8 High-dose melphalan with stem cell transplant (SCT) is also a standard frontline therapy. High rates of hematologic and organ response have now been documented at multiple centers, with long-term data reported on over 800 patients and median survivals of over a decade for SCT patients achieving complete response.25,26 Enthusiasm for SCT has been tempered by the high treatment-related mortality, which remains around 5% to 10% even at experienced centers.25,26 Risk-adapted SCT, which tailors the dose of melphalan according to the age and risk status of the patient, may improve early survival, with a treatment-related mortality of 4% in a recent phase II study.17 To compensate for the loss of efficacy related to attenuated conditioning, adjuvant therapy post-SCT for patients not achieving a CR has been tested and shown to improve hematologic response. Both thalidomide + dexamethasone (TD) and bortezomib + dexamethasone (BD) have been studied as adjuvant therapy post-SCT. CR rates at 12 months post-SCT were achieved in 39% and 65% of evaluable patients, respectively, in these two studies, with high rates of organ improvement.17,19 An updated analysis of survival on the adjuvant TD trial showed 69% of patients alive with a median follow-up of 52 months.27
Despite such success, only a quarter of newly diagnosed AL patients are candidates for melphalan at 140 or 200 mg/m2. Indeed, the outcomes reported above with SCT reflect selection bias—that is, choosing the fittest patients for SCT. The two frontline therapies, MDex and SCT, were compared by the French Myélome Autogreffe Groupe in a multicenter, randomized, phase III trial in newly diagnosed AL patients. There were no significant differences in hematologic or organ responses, but median OS was significantly better in the MDex arm (56.9 vs. 22.2 months, p = 0.04).24 However, 22 of the 50 patients assigned to SCT (44%) were not evaluable for response or long-term survival, including 13 who never received SCT due to death or progression and nine who died peri-SCT. Although the updated event-free survival of patients in both arms of the study continued to show equivalence (Figure 2D), confirming the importance of MDex in the treatment of AL, the small number of evaluable SCT patients in this phase III trial, the disparity between outcomes in the SCT arm and SCT results at experienced centers, and the lurking risk of myelodysplasia and secondary leukemia with oral melphalan (a risk reduced if the total dose is kept at < 600mg) make it difficult to conclude that equivalence means that MDex is best for all patients.
Improved Outcomes with Novel Agents
Over the past decade, we have witnessed improved survival for many patients because of new drugs being approved for myeloma. In contrast, clinical research in AL has been limited to small, investigator-initiated trials that are usually without benefit of phase I dose-finding components, likely indicating that AL has not been a primary focus because of its rarity. Additional important reasons for the lack of drug development for AL include the propensity for sudden cardiac death, the frequency of multi-organ involvement, and the consequent potential confounding of progressive organ disease and drug-related side effects. Until recently, a biomarker-based cardiac-staging system and a framework for objectively scoring hematologic and organ responses were not available.
Regimens and results from a selection of trials with novel agents in AL are shown in Table 2. The first novel agent to be investigated in relapsed AL, thalidomide, was poorly tolerated at high doses but showed efficacy at moderate doses in combination with dexamethasone, with hematologic responses in 40% to 50% of patients post-SCT.17,28 The combination of oral cyclophosphamide + thalidomide + dexamethsone (CTD) showed promise in the relapse setting and in a single-center retrospective series of 122 newly diagnosed patients (48% with heart involvement). CTD had high hematologic response rates, with a median time to response of 2 months and 74% survival at 3 years.29 (A recent case-control comparison of CTD and MDex in AL showed no significant differences in outcomes between the groups.30 ) CTD is stem-cell sparing, unlike MDex, but the side effects of thalidomide, particularly neuropathy, bradycardia, and worsening congestive heart failure, remain significant in AL.
Selected clinical studies of novel agents in AL amyloidosis
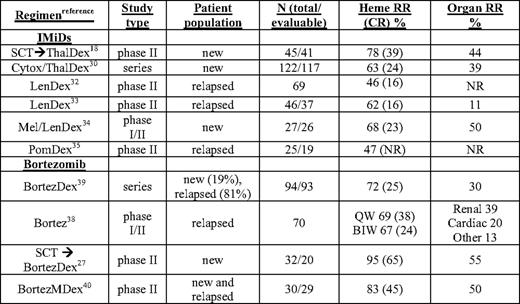
BIW, twice weekly bortezomib schedule; Bortez, bortezomib; CR, complete response rate; Cytox, cyclophosphamide; dex, dexamethasone; Len, lenalidomide; mel or M, melphalan; new, untreated patients; NR, not reported; Pom, pomalidomide; QW, weekly bortezomib schedule; RR, response rate; SCT, autologous stem cell transplant; series, retrospective series; Thal, thalidomide
In phase II trials, full-dose lenalidomide had significant toxicity requiring dose reductions or discontinuation, and was better tolerated at 15 mg/d in combination with weekly dexamethasone (LenDex), with hematologic response rates of 40% to 50%.31 In recent reports of more than 100 patients with relapsed AL treated with LenDex, there was a 52% hematologic response rate with 16% CR, median OS of about 2 years, and, in those achieving CR, PFS of over 3 years.32,33 Several phase I/II studies combining LenDex with oral melphalan or cyclophosphamide are ongoing, although preliminary indications are that dose reductions of the myelosuppressive agents are required and response rates are not substantially different from those seen with MDex.34 In a preliminary report on a phase II study of pomalidomide with weekly dexamethasone (PomDex), over one-third of heavily pretreated relapsed AL patients achieved a hematologic response by 6 months, highlighting the promise of this potent immunomodulatory drug (IMiD).35 Maintenance therapy with IMiDs eventually may prove important in AL as well as myeloma. A notable confounding aspect of IMiD therapy in AL is a rise in cardiac biomarkers that does not correlate with worsening cardiac status or hematologic progression. In one trial, asymptomatic increases in NT-proBNP by more than 30% (>300 ng/L) occurred in 67% of patients after cycle 1.36 This represents yet another challenge to drug testing in AL: the confounding effect on metrics of organ involvement and response. In such circumstances, it is advisable to assess organ responses after a drug holiday or washout period.
The safety and preliminary efficacy of bortezomib as a single agent in relapsed AL has been evaluated in a recent phase I study of 31 patients.37 The maximum tolerated dose was not reached, and further evaluation of dosing at both 1.3 mg/m2 on the standard schedule (biweekly, BIW) and 1.6 mg/m2 when given weekly (QW) for 4 of 5 weeks was completed on the phase II level. In this phase I/II trial (N = 70), the most common side effects were gastrointestinal, with grades 3 and 4 vomiting and diarrhea in five patients (adjusted prophylaxis with loperamide and granisetron was routinely used in QW patients).38 In the QW cohort (N = 18), there were no instances of grade 3 or above peripheral neuropathy and no discontinuations due to peripheral neuropathy. Hematologic response (ORR) and CR rates were among the highest ever reported for a single agent (CR rates appeared to be dose related): 69% ORR with 38% CR QW, 67% ORR with 24% CR BIW (n = 34), and 39% ORR with 11% CR at lower doses. PFS at 1 year exceeded 75% and OS 90%.38 Renal responses were seen in 14/49 (29%), and objective cardiac responses in 5/39 (13%).
In a retrospective series of 94 patients (81% previously treated) receiving bortezomib BIW and dexamethasone, there was a 71% ORR with 25% CR, a median time to progression of 25 months, and a 1-year survival rate of 75%.39 In a phase II clinical trial, bortezomib and dexamethasone adjuvant therapy for patients not achieving a CR post-SCT resulted in over 90% of patients improving their hematologic responses. At 1 year post-SCT, 65% had achieved a CR and 55% had organ responses.19 Most striking in the clinical trials and case series in which AL patients received bortezomib was the rapid time to response, typically about 1 month, a critical variable in patients with end-organ damage.
Bortezomib is currently being evaluated in combination with melphalan and dexamethasone (BMDex), with a best-response rate of over 80% and a CR rate of 42%.40 In the first 35 patients treated in that phase II trial, 40% required dose reductions of bortezomib for thrombocytopenia or peripheral neuropathy, but only one patient was removed from the study for grade 3 peripheral neuropathy. These data, along with the improved survival with bortezomib and oral melphalan and prednisone in myeloma, justify the comparison of BMDex to MDex for frontline therapy in AL in phase III trials planned by the Eastern Cooperative Oncology Group and a network of centers in Europe. The basis for the impressive activity of bortezomib in AL may be that AL plasma cells are exquisitely sensitive to proteasome inhibition due to the fitness costs exacted by light-chain misfolding or aggregation on both the protein-folding environment and the capacity of endoplasmic reticulum-associated degradation pathways.41
Combination Therapies and a Framework for Drug Development for Systemic AL Amyloidosis
As encouraging as these results are, given the activity of IMiDs and bortezomib in AL, it is disappointing that there has not been support for a clinical trial combining these agents for the treatment of an AL study population. The rapid disease control provided by bortezomib and the durability of CR with long-term IMiD therapy argue strongly for such a trial. This would require a phase I component and, if dexamethasone were included, a three-drug dose-reduction schema. There is clearly a need for a proactive AL clinical research network, for foundation support and CTEP guidance, and for a drug-development framework for AL that nurtures commitment.
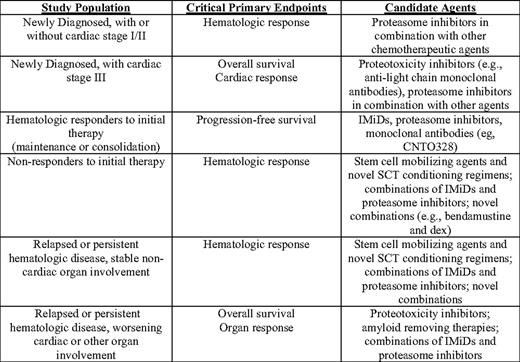
The list of agents active or in testing for myeloma includes (to name only a few) bendamustine, monoclonal antibodies (IMGN901, CNTO 328, Elotuzumab, MF4809g), histone deacetylase and Hsp90 inhibitors, and new proteasome inhibitors, including irreversible (Carfilzomib) and oral (MLN 9708) formulations. Not all of these agents will have activity alone or in combination in AL. A clinical research network is needed for AL patients in order to perform (and make widely available) the phase I/II trials required for evaluation of novel or combination therapies. Such a network would provide pharmaceutical companies with the nexus for testing new agents in AL as the road ahead for new agents for myeloma becomes grid-locked, and could channel the most promising concepts to the National Cancer Institute and cooperative groups for consideration for randomized phase II and III trials. One hopes that among the future agents tested for AL will be novel drugs that act to inhibit proteotoxicity (the toxic effects of misfolded light chains and aggregates on cells) analogous to the amyloid inhibitors that continue to be developed for Alzheimer's disease. The framework for clinical research in AL can be more clearly apprehended today than a decade ago, particularly with the validation of cardiac risk-assessment staging, the development of hematologic and organ-response criteria, and the longer survival of more patients from the time of diagnosis. We suggest such a framework in Table 3. A critical feature is that the progression of organ disease and imminence of death are most salient in two categories of patients: those newly diagnosed with stage III cardiac disease and those with relapsed AL and worsening organ involvement. With respect to the latter, the tempo of organ failure can accelerate late in the course of disease despite minimally measurable iFLC or M proteins due to increased foci of deposition and declining reserve in involved organs.
A major challenge moving forward is the standardization of adverse-event reporting. The phase I/II trial of bortezomib provides a template in this regard. Patient disposition with each cycle and all categories and grades of adverse events occurring in 10% or more patients were tabulated based on investigator reporting with each cycle. Such a systematic prospective approach to the details of each patient's course is a particularly useful and transparent way to minimize confounding of progressive amyloid-related organ disease and drug-related side effects. It helped that bortezomib had such prompt activity against the monoclonal disease; in other instances, adverse-event monitoring and reporting may be more difficult despite the best-laid plans.
Conclusions: Seeing the Future
The past decade has seen significant advances in the treatment of patients with AL, leading to improvements in both quality of life and survival. Unfortunately, however, patients continue to present with advanced and often irreversible organ failure, emphasizing the continued need for education to promote earlier diagnosis and for alternative approaches, including solid organ transplantation in young patients. The availability of novel agents has significantly expanded our armamentarium against AL, and the central challenges of this decade will be how to combine these agents and how to bring forth new ones for approval. A working framework of response and end-point criteria and standardization of adverse event reporting will accelerate the transition to serious drug development and testing for AL. Over this decade, it is also likely that SCT will become more appropriate as a second-line therapy, creating new issues for stem-cell mobilization and conditioning regimens. Hopefully, with continued national and international collaboration and patient advocacy, many of these questions can be defined and addressed, leading to improved outcomes for future patients with AL.
Disclosure
Conflict-of-interest disclosure: RLC has been a consultant for Elan Pharmaceuticals, has received research funding from Genzyme, Celgene, and Ortho, and also has a membership on the board of directors/advisory committees for Millennium Pharmaceuticals. ADC has received honoraria and is associated with the speakers' bureau of Celgene and Millennium Pharmaceuticals. Off-label drug use: None disclosed.
Correspondence
Raymond L. Comenzo, MD, Tufts Medical Center, Box 826, 800 Washington St., Boston, MA, 02111; Phone: 617–636–6454; Fax: 617–636–3175; e-mail: rcomenzo@tuftsmedicalcenter.org
