
Abstract
The contribution of platelets to normal hemostasis and vascular disease is well described. However, recent studies make it clear that much remains to be learned about platelet activation at the single cell and the molecular level, and about the contribution of platelets to inflammation, tumor angiogenesis, and embryonic development. This article is divided into two themes. The first is an overview of current knowledge of the mechanisms that drive platelet function in vivo and a brief summary of some of the emerging ideas that are modifying older views. The second theme is a consideration of the strengths and weaknesses of the tools we have as hematologists to assess platelet function in the clinical setting, identify mechanisms, and evaluate the impact of antiplatelet agents.
Although fish, birds, and presumably dinosaurs make do with nucleated thrombocytes as the cellular component of hemostasis, platelets evolved in mammals as a specialized means to seal leaks in a high-pressure, high-flow circulatory system. Many of the most pertinent properties of platelets—including their shape, the contents of their secretory granules, their high density of regulatable adhesion receptors, and their ability to promote thrombin generation—are dictated by the requirements of forming a stable hemostatic plug under high-flow conditions. Circulating platelets must be able to sustain repeated contact with the normal vessel wall without premature activation, recognize the unique features of a damaged wall, cease forward motion upon recognition of damage, adhere despite the forces produced by continued blood flow, and cohere to each other, forming a stable plug of the correct size that can remain in place until it is no longer needed. This brief review considers some of the current ideas about platelet activation and the evaluation of platelet function in the research and clinical settings.
Overview of Platelet Activation
The contribution of platelets to hemostasis is different in arteries and veins. In the venous system, low-flow rates and stasis permit the accumulation of activated coagulation factors and the local generation of thrombin largely with a less prominent contribution from platelets. Venous thrombi contain platelets, but the dominant cellular component consists of trapped erythrocytes. In the arterial circulation, higher flow rates limit fibrin formation by washing out soluble clotting factors. Platelets, which work best at higher shear rates, help to form a physical barrier against further blood loss and, at the same time, provide a surface on which thrombin is generated and fibrin can accumulate. Activated endothelial cells participate in this process as well, promoting fibrin deposition by helping to assemble activated clotting factor complexes, but also releasing prostaglandin I2 and nitric oxide, which inhibit platelet function, and promoting activation of protein C, which inhibits thrombin formation by cleaving factors Va and VIIIa.
Pathologic thrombus formation in the arterial system occurs when diseases or drugs subvert the mechanisms intended to prevent unwarranted platelet activation, producing a local accumulation of platelets where none was needed. Pathologically high levels of shear can activate platelets directly, but, in general, platelets are activated by agonists whose receptors are expressed on the platelet surface. The extent of the platelet response to injury is subject to tight regulation. Appropriate platelet activation limits the extent of blood loss following vascular injury and promotes subsequent wound healing without causing vascular occlusion. Inappropriate platelet activation, whether initiated by plaque rupture or less dramatic vascular events, leads to arterial occlusion, ischemia, and tissue injury.
In the setting of the hemostatic response to injury, platelets are activated by collagen and thrombin. Collagen fibrils within the vessel wall become exposed to the circulation when the endothelial cell monolayer is breached, forming a complex with von Willebrand factor (VWF). Platelets tumbling on the periphery of the rapidly moving bloodstream are captured when glycoprotein (GP) Ibα on the platelet surface binds to the VWF A1 domain, establishing contacts that slow the forward progress of the platelet long enough for platelet activation to occur. The higher shear rates present in arteries help to expose the A1 domain in anchored VWF multimers. Drivers for platelet activation include the signaling events that occur downstream of receptors for collagen (GP VI and GP Ibα), thrombin (PAR1 and PAR4), adenosine diphosphate (ADP; P2Y1 and P2Y12), and thromboxane A2 (TxA2; TP). Erythrocytes facilitate this process in part by pushing platelets closer to the vessel wall and in part by providing a source of ADP following injury. Human platelets express 15,000 to 25,000 copies of GP Ibα per cell, a high density for a cell as small as a platelet. GP Ibα exists as part of a multiprotein complex with GP Ibβ, GP IX, and GP V, in which GP Ibα stands out from the platelet surface ready to engage VWF on the vessel wall. Mutations in the GP Ib complex that prevent its expression on the platelet surface or that impair GP Ibα receptor function produce a bleeding disorder (Bernard-Soulier syndrome) because platelet adhesion to the vessel wall is impaired. Macrothrombocytopenia arises from a failure to form the normal linkages between GP Ibα complex and filamin in the platelet membrane cytoskeleton.
A Conventional View of the Stages in Platelet Activation.
The formation of a stable platelet plug following vascular injury is often described as occurring in three distinct stages: (1) initiation, (2) extension, and (3) stabilization (Figure 1). Initiation by collagen or thrombin produces a platelet monolayer that supports the subsequent adhesion of activated platelets to each other. Extension occurs when additional platelets adhere to the initial monolayer and become activated. Thrombin, ADP, and TxA2 play an important role in this step, activating platelets via cell surface receptors coupled to heterotrimeric G proteins (Figure 2). ADP is secreted from storage sites within platelet-dense granules. TxA2 is synthesized from arachidonic acid released from platelet membrane phospholipids when platelets are activated. TxA2 formation in platelets is dependent on cyclooxygenase-1 (COX-1), an enzyme that is irreversibly inhibited by aspirin and reversibly inhibited by most other nonsteroidal antiinflammatory agents. The local generation of thrombin is facilitated by activated platelets that provide a surface on which clotting factor complexes can be assembled once phosphatidylserine has moved to the platelet surface from the inner leaflet of the plasma membrane. Intracellular signaling downstream of agonist receptors activates integrin αIIbβ3 (GP IIb-IIIa), making cohesive interactions between platelets (ie, aggregation) possible.

Stages in platelet plug formation. A classical model. (A) Prior to vascular injury, platelet activation is suppressed by endothelial cell-derived inhibitory factors. These include prostaglandin PGI2 (prostacyclin), nitric oxide (NO), and CD39, an ADPase on the surface of endothelial cells that can hydrolyze trace amounts of ADP that might otherwise cause inappropriate platelet activation. (B) Initiation. The development of the platelet plug is initiated by thrombin and by the collagen-VWF complex, which captures and activates moving platelets. Platelets adhere and spread, forming a monolayer. (C) Extension. The platelet plug is extended as additional platelets are activated via the release or secretion of TxA2, ADP, and other platelet agonists, most of which are ligands for G protein-coupled receptors on the platelet surface. Activated platelets stick to each other via bridges formed by the binding of fibrinogen, fibrin, or VWF to activated αIIbβ3. (D) Stabilization. Finally, close contacts between platelets in the growing hemostatic plug, along with a fibrin meshwork (shown in red), help to perpetuate and stabilize the platelet plug. This model is being revised as new observations (described in the text) of the behavior of individual platelets within the hemostatic plug add additional refinements.
Stages in platelet plug formation. A classical model. (A) Prior to vascular injury, platelet activation is suppressed by endothelial cell-derived inhibitory factors. These include prostaglandin PGI2 (prostacyclin), nitric oxide (NO), and CD39, an ADPase on the surface of endothelial cells that can hydrolyze trace amounts of ADP that might otherwise cause inappropriate platelet activation. (B) Initiation. The development of the platelet plug is initiated by thrombin and by the collagen-VWF complex, which captures and activates moving platelets. Platelets adhere and spread, forming a monolayer. (C) Extension. The platelet plug is extended as additional platelets are activated via the release or secretion of TxA2, ADP, and other platelet agonists, most of which are ligands for G protein-coupled receptors on the platelet surface. Activated platelets stick to each other via bridges formed by the binding of fibrinogen, fibrin, or VWF to activated αIIbβ3. (D) Stabilization. Finally, close contacts between platelets in the growing hemostatic plug, along with a fibrin meshwork (shown in red), help to perpetuate and stabilize the platelet plug. This model is being revised as new observations (described in the text) of the behavior of individual platelets within the hemostatic plug add additional refinements.

Pathways that support platelet activation. A summary of some of the major signaling pathways and events of platelet activation. Further details in the text. PLC, phospholipase C; PKC, protein kinase C; IP3, inositol-1,4,5-trisphosphate; αIIbβ, refers to the platelet integrin that is also known as GP IIb-IIIa in older literature; IP and TP, PGI2 and TxA2 receptors.
Pathways that support platelet activation. A summary of some of the major signaling pathways and events of platelet activation. Further details in the text. PLC, phospholipase C; PKC, protein kinase C; IP3, inositol-1,4,5-trisphosphate; αIIbβ, refers to the platelet integrin that is also known as GP IIb-IIIa in older literature; IP and TP, PGI2 and TxA2 receptors.
As previously noted, most of the agonists that extend the platelet plug do so via G protein-coupled receptors. The properties of these receptors make them particularly well suited for this task. Most G protein-coupled receptors bind their ligands with high affinity and, because they act as exchange factors, each occupied receptor can theoretically activate multiple G proteins. This allows amplification of a signal that might begin with a relatively small number of receptors. Binding studies show that agonist receptors are expressed on the platelet surface in low copy number, ranging from a few hundred (epinephrine receptors and P2Y1 ADP receptors) to a few thousand (PAR1 and GP VI) copies per cell. Because mechanisms exist that can limit the activation of G protein-coupled receptors, platelet activation can be tightly regulated, even at its earliest stages. In this context, the impact of the family of RGS (regulator of G protein signaling) proteins in platelets is just beginning to be explored. These are proteins that, in cells other than platelets, have been shown to limit signaling intensity and duration by accelerating the hydrolysis of GTP (guanosine-5′-triphosphate) by activated G protein α subunits.1 They may play a similar role in platelets. Although the total number of G protein-coupled receptor genes expressed in platelets still remains to be determined, platelets have been shown to express at least 10 different G proteins, and distinct roles have been established for many of them in mice.2
Gaps in the Map.
Despite considerable effort, gaps remain in signaling maps that attempt to connect receptors on the platelet surface with the most characteristic platelet responses, although the multiplicity of known signaling molecules and pathways may tend to obscure that (Figure 2). For the most part, platelet activation begins with the activation of a phospholipase C (PLC) isoform, which by hydrolyzing membrane phosphatidylinositol-4,5-bisphosphate produces the second messengers needed to raise the cytosolic Ca2+ concentration, leading to integrin activation via a pathway that includes an exchange factor (CalDAG-GEF), a switch (the Ras family member Rap1), an adaptor (Rap1-GTP-interacting adaptor molecule [RIAM]), and proteins that interact directly with the integrin cytosolic domains (kindlin and talin) (Figure 2). Which isoform of PLC is activated depends on the agonist. Collagen activates PLCγ2 using a mechanism that depends on scaffold molecules and protein tyrosine kinases. Thrombin, ADP, and TxA2 activate PLCβ using Gq as an intermediary.
The rise in cytosolic Ca2+ that occurs during platelet activation is thought to occur as a two-step process. The first is the inositol-1,4,5-trisphosphate-mediated release of Ca2+ from the platelet-dense tubular system. This triggers an influx of Ca2+, driven by the steep Ca2+ concentration gradient that normally exists across the plasma membrane. This process requires two proteins: (1) STIM1 in the dense tubular system and (2) Orai1 in the plasma membrane.3 The rise in Ca2+ leads to activation of Rap1 mediated primarily by the Ca2+-sensitive exchange factor, CalDAG-GEF. Rap1 can then bind to and cause the translocation of RIAM and talin.
Talin binding to the cytoplasmic domain of αIIbβ3 is thought to trigger a conformational change in the integrin, exposing a binding site for fibrinogen.4 The binding of bivalent fibrinogen makes it possible for platelets to adhere to one another. Other proteins that can substitute for fibrinogen include fibrin, VWF, and fibronectin. Average expression levels of αIIbβ3 range from approximately 50,000 per cell on resting platelets to 80,000 on activated platelets, translating into a remarkably high density of expression for a cell as small as a platelet. Mutations in αIIbβ3 that prevent expression or abolish function produce a bleeding disorder (Glanzmann's thrombasthenia) because platelets are unable to form the stable aggregates required for hemostasis. To a more limited extent, a similar process occurs in patients receiving integrin-blocking agents.
“Stabilization,” the third stage of platelet plug formation illustrated in Figure 1, refers to the later events of platelet plug formation that help to stabilize the platelet plug and prevent premature disaggregation, in part by amplifying signaling within the platelet. Examples include outside-in signaling through integrins and signaling through receptors whose ligands are located on the surface of adjacent platelets (Figure 2). The net result is a hemostatic plug comprised of activated platelets embedded within a cross-linked fibrin mesh, a structure stable enough to withstand the shear forces generated by flowing blood in arterial circulation. In the process, platelets not only provide a surface that can facilitate leukocyte emigration into surrounding tissues, but can also serve as a source of inflammatory mediators and molecules that promote wound healing.
The model of platelet activation summarized in Figure 1 arises from studies on platelets from individuals with monogenic disorders of platelet function and from mouse models. Many of the most critical molecules have been knocked out in mice. Examples of proteins that are affected by well-characterized gene defects in humans include αIIbβ3 (Glanzmann's thrombasthenia), GP Ib (Bernard-Soulier syndrome), VWF (von Willebrand's disease), and the collagen receptor, GP VI, among others.5 Understanding platelet function at this level provides a framework for the development of antiplatelet agents. In general, the antiplatelet agents that are in current clinical use can be divided into three categories. (1) Drugs that inhibit platelet activation by blocking the receptors for agonists such as ADP and, more recently, thrombin. Clinically available ADP P2Y12 receptor antagonists include Clopidogrel and Prasugrel. Antagonists for the PAR1 thrombin receptor are in clinical trials. (2) Drugs that inhibit platelet activation by blocking intracellular signaling or raising platelet cAMP (cyclic AMP) levels. Examples include aspirin (which prevents TxA2 formation by inhibiting COX-1) and dipyridamole (which retards cAMP hydrolysis by inhibiting cAMP phosphodiesterase). (3) Drugs that prevent activated platelets from aggregating by blocking αIIbβ3. Examples include Abciximab (ReoPro), tirofiban (Aggrastat), and eptifibatide (Integrilin) (Figure 2).
Emerging Ideas About Platelets and Platelet Function
Platelets Are Not Just for Hemostasis Any More.
Some of the most interesting recent work on platelet biology has to do with the discovery of new roles for platelets. The results have clinical, as well as therapeutic, implications. Thus, it has recently been shown that that platelets participate in primary immunity by helping to trap bacteria in neutrophil-derived DNA nets, affect tumor progression by stimulating angiogenesis, help to close the ductus arteriosus at the time of birth, contribute inflammatory mediators in some forms of arthritis, and play a role in the separation of the lymphatic circulation from the arterial and venous circulation during embryogenesis. A recent commentary brings many of these observations together.6
Spatial and Temporal Relationships.
Recent observations indicate that the model shown in Figure 1 is oversimplistic, because it suggests that platelet accumulation after injury is an orderly series of events. In fact, there is now ample evidence that there is spatial, as well as temporal, heterogeneity within a growing hemostatic plug and that the attachment of new platelets is transient unless a sufficient degree of activation of that particular platelet is achieved.7–9 Historically, the challenge has been to explain how fast-moving platelets can linger long enough to become activated and form stable attachments to the growing mass. Platelets incorporated into the thrombus can release soluble molecules that act as recruiting factors, transforming nearby platelets into the activate state. However, even assuming that the local concentration of these molecules becomes high enough to work, by the time freely moving platelets can respond, they would have moved downstream of the site of injury.
Newer research shows that platelets slow down in part by forming membrane tethers, bypassing the need to be activated on arrival. In addition, published reports10 and studies that we have performed show that the platelet mass that accumulates after laser injury is heterogeneous. A core of fully activated platelets is overlaid with platelets that are not yet positive for the α-granule membrane protein, P-selectin, whose appearance on the platelet surface serves as a convenient marker that secretion has occurred. Initially, platelets form a thrombus without turning P-selectin positive. With time, the small, inner core of platelets becomes P-selectin+, a property that then spreads outward. Meanwhile, the overlying shell of P-selectin– platelets includes transition zones in which platelets can become attached. Thus, platelet activation and accumulation have spatial, as well as temporal, diversity. Understanding how the transitions between states are regulated may provide clues for the development of new classes of antiplatelet agents.
Contact-dependent Signaling.
Formation of either a hemostatic plug or a pathological thrombus has the net effect of bringing platelets into close contact with each other. Electron micrographs of aggregated platelets show the close proximity of adjacent platelets, allowing molecules on the surface of adjoining platelets to interact and pass information, much like a neurological or immunological synapse. This can be an indirect interaction, such as occurs when fibrinogen bridges activated αIIbβ3 on adjacent platelets, or a direct interaction, as when one cell adhesion molecule binds to another in trans. Examples of the latter include interactions mediated by members of the CTX (cortical thymocyte marker in Xenopus) family of adhesion molecules.11 Junctional molecules are expressed in platelets, and there is increasing evidence that biologically meaningful interactions occur and that some platelet surface molecules accumulate at sites of contact.12,13
Direct contacts between platelets can promote signaling by more than one mechanism. In addition to signaling events that occur downstream from cell adhesion molecules, there are receptors that can interact with cell surface ligands on adjacent platelets once the platelets have come into sufficiently stable contact with each other. One example is the family of Eph receptor tyrosine kinases and their ligands, known as ephrins. Ephrins are cell surface proteins. Contact between an ephrin-expressing cell and an Eph-expressing cell allows bidirectional signaling. Human platelets express at least EphA4, EphB1, and ephrinB1. Blockade of Eph/ephrin interactions leads to reversible platelet aggregation and limits the growth of platelet thrombi on collagen-coated surfaces.14 A second example is the binding of the semaphorin family member, sema4D (CD100), to its receptors. Sema4D is an integral membrane protein with a cytoplasmic domain. Signaling downstream of sema4D receptors promotes platelet activation by collagen. Loss of sema4D expression in mice inhibits platelet function in vitro and thrombus formation in vivo.15,16
Shedding and Secreting to Communicate.
In addition to forming an essential part of the hemostatic plug, platelets are delivery vehicles, secreting proteins from their α-granules and small molecules from their dense granules.17 These molecules have numerous established and imputed roles, including helping to activate additional platelets, promoting platelet adhesion and aggregation, reducing blood loss by causing vasoconstriction and, eventually, promoting wound healing. An additional class of platelet-derived proteins arises by ectodomain shedding by metalloproteases.18 In theory, ectodomain shedding can serve multiple roles, including modulating adhesive and cohesive interactions by and between platelets, limiting platelet responses to agonists, and enabling cryptic functions of the cleaved proteins. Shedding can also provide a source of bioactive fragments that bind to receptors on other cells, modifying their behavior. GP Ibα and GP VI are examples of platelet receptors that are subject to proteolytic shedding. Surface proteins that provide a source of bioactive fragments include CD40L, P-selectin, and sema4D. However, there are also proteins shed from platelets whose role in platelet biology, either before or after shedding, is not entirely clear. The amyloid β precursor protein is an example. At a practical level, the appearance of soluble P-selectin and CD40L has been used in clinical studies to track platelet activation in vivo, and the interaction of CD40L with its receptors is a target for drug development.
Evaluating Platelet Function In Vitro and In Vivo
In some respects, the approaches used for platelet function testing in the clinical and research settings have diverged, largely because many of the technologies now routinely applied in the research laboratory have either not been translated into technologies that can be used for studies on people or have not found homes in clinical laboratories, wherein the results can be used to support clinical decision making. There are important exceptions; but, in general, efforts on the clinical side have focused on the development of commercial point-of-care devices that can be used close to the patients bedside by a minimally trained observer. These devices are intended more for the monitoring of patients on antiplatelet agents than for the evaluation of patients with bleeding disorders.
One of the oldest technologies of all, light transmission aggregometry and its more recent iterations—lumi-aggregometry and whole blood aggregometry—have remained as useful in the diagnosis of some well-characterized hereditary platelet disorders in humans because they are in the study of platelets from transgenic mice. However, platelet behavior in the low shear environment of an aggregometer cuvette does not necessarily reflect all aspects of platelet behavior in vivo. Flow cytometry using antibodies that recognize proteins on the platelet surface or the phosphorylated forms of intracellular proteins has moved from the research laboratory to the specialty clinic, but widely used research methods that allow the behavior of individual platelets to be observed under flow in vivo or in vitro have not yet made the jump.
Assessing Platelet Function in the Research Setting.
Some of the current methods to study platelet function are summarized in Table 1 and Table 2. In the research setting (Table 1), an important goal has been to develop better methods to observe platelet function in vivo so that hypotheses can be tested. Mice have become the model animal of choice in part because they can be genetically manipulated by design. For example, it is now possible to use digital confocal fluorescence intravital microscopy in mice to observe the behavior of individual platelets being recruited into a hemostatic plug following vascular injury.19 This and related approaches have proved to be especially useful in uncovering mechanisms of platelet activation. However, for the most part, these approaches are limited to the microcirculation in accessible places, such as the mesentery and cremaster muscle; locations in which the vessel wall is thin enough to allow direct observation of events within the lumen. Platelet activation in larger arteries is commonly studied after inducing an oxidative injury with FeCl3 or rose Bengal, acute interventions that are not especially physiologic. Because the wall of the larger arteries is too thick to permit detailed observations of individual platelets, a common, but much less satisfactory, endpoint is the time required to form an occlusive thrombus. These models are very useful, especially when recording the impact of gene deletions and substitutions, but they are inherently limited because of differences between mice and humans.
Evaluating platelet function in the research setting
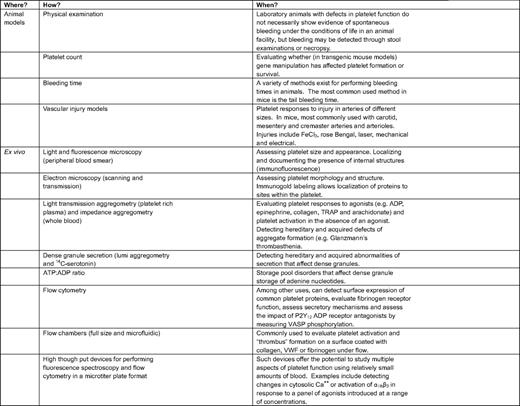
ATP indicated adenosine triphosphate; PAR-1, protease-activated receptor-1; TRAP, thrombin receptor activation peptide (an agonist for platelet PAR-1); VASP, vasodilator-stimulated protein.
Evaluating platelet function in the clinical setting
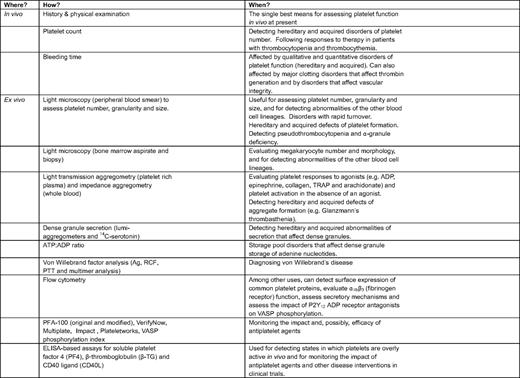
ATP indicates adenosine triphosphate; Ag, antigen; RCF, ristocetin cofactor; PTT, partial thromboplastin time; TRAP, thrombin receptor activation peptide (an agonist for platelet PAR-1); VASP, vasodilator-stimulated protein.
An alternative approach that is applicable to studies of human platelets is to pump or pull whole blood, platelet-rich plasma, or washed platelets through a flow chamber coated with collagen, fibrinogen, or VWF. Such devices have the advantages of allowing platelet behavior to be studied under flow, more closely mimicking this aspect of arterial conditions. Recent progress with microfluidic devices has made it possible to scale down the size of flow chambers, greatly reducing the amount of blood required—an obvious advantage for studies on mouse platelets—but also foretelling the development of standardized devices that might eventually be useful in the clinical setting.20
Assessing Platelet Function in the Clinical Setting.
In the clinical setting, platelet function tests are most commonly either a diagnostic aid, an adjunct to decision making, or both. Typical questions that hematologists ask include: Will this patient bleed if taken to surgery? If bleeding occurs, what should be done to make it stop? Does this patient have a well-characterized disorder that has predictable impact on hemostasis or thrombosis? Cardiologists, in contrast, are more likely to ask whether an optimal dose of an antiplatelet agent is being administered to help avoid secondary events or prevent stent thrombosis.
Figure 3 shows four common uses for platelet function testing in the clinical setting: (1) determining whether there is an intrinsic defect that impairs platelet function; (2) documenting whether platelets are more reactive than they should be in vivo; (3) identifying possible mechanisms that will aid in diagnosis and decision making; and (4) helping to assess the impact of antiplatelet agents, especially those that target platelet P2Y12 ADP receptors. Each of these is considered briefly below with citations and references that consider the topic in greater detail than space and time allow here. Table 2 summarizes some of the methods that are currently available.

Platelet function testing in the clinical setting is usually performed in a specialized laboratory where sufficient numbers of patients can be studied on an annual basis to ensure reproducibility and correct interpretation of the results. Although there are clearly exceptions, platelet function tests are usually performed either to determine the cause of bleeding in a nonthrombocytopenic patient with normal coagulation parameters or to assess the impact of antiplatelet agents, such as aspirin, the P2Y12 ADP receptor antagonists. With the exception of von Willebrand's disease, most hereditary platelet disorders are rare. Leaving aside patients receiving drugs that affect platelet function, a well-characterized mechanism is identified less than half the time.
Platelet function testing in the clinical setting is usually performed in a specialized laboratory where sufficient numbers of patients can be studied on an annual basis to ensure reproducibility and correct interpretation of the results. Although there are clearly exceptions, platelet function tests are usually performed either to determine the cause of bleeding in a nonthrombocytopenic patient with normal coagulation parameters or to assess the impact of antiplatelet agents, such as aspirin, the P2Y12 ADP receptor antagonists. With the exception of von Willebrand's disease, most hereditary platelet disorders are rare. Leaving aside patients receiving drugs that affect platelet function, a well-characterized mechanism is identified less than half the time.
Determining Whether There Is an Intrinsic Defect That Impairs Platelet Function.
Many of the hematologists who I have encountered, myself included, believe that the very best test of hemostatic function in people is a detailed history and physical examination performed by an experienced observer, particularly in individuals who have been hemostatically challenged by surgery, trauma, or the vicissitudes of daily life. However, mucocutaneous bleeding can arise from more than just platelet defects, because it reflects the integrity of the vasculature and the presence of critical plasma proteins, as well as quantitative and qualitative platelet defects. The most common hereditary disorder of platelet function is VWF's disease, which is not a platelet defect at all. The first goal of platelet function testing in the clinical laboratory is, therefore, to document that an intrinsic function defect may exist and exclude VWF deficiency. The bleeding time is theoretically helpful in this process; but, because it is subject to defects extrinsic to the platelet, can cause scarring and is not reliable for predicting clinical outcomes, measurement of the bleeding time has fallen out of favor, at least in humans.21 If suspected, von Willebrand's disease should be diagnosed or excluded by specific tests of VWF expression and function, including VWF (antigen), ristocetin cofactor activity, factor VIII levels, and agarose gel electrophoresis to assess VWF multimer formation.
Despite its age, nothing has fully supplanted platelet aggregometry for documenting that platelets are not working properly. Yet, whether it is done in a standard light transmission aggregometer, a lumi-aggregometer (allowing simultaneous detection of dense granule secretion), or an impedance (whole blood) device, aggregometry remains as much an art as a science. Aggregation studies are subject to poor reproducibility, which can be reduced, but not completely eliminated, with operator experience and careful attention to factors (eg, the anticoagulant that is used, the time of day and temperature when the blood is obtained, and the time elapsed between drawing the blood and running the assays).22,23 Even when performed under optimal conditions, aggregometer studies are not necessarily predictive of bleeding risk, although they can be when combined with a detailed clinical history.22–26 Aggregometry is most predictive of outcome when abnormalities are detected that lead to the diagnosis of a well-defined molecular defect whose clinical implications are thoroughly understood.24 Lumi-aggregometry offers the additional ability to measure dense-granule secretion in real time along with platelet aggregation. It is less evident that newer devices, such as the PFA-100,27 can or should replace aggregometry for determining whether a platelet function disorder is present, even though they offer greater ease of performance.28 The use of thromboelastography to evaluate platelet function and the impact of antiplatelet agents requires further study.29–31
Documenting Whether Platelets Are More Reactive Than They Should Be In Vivo.
The converse of evaluating platelet function defects in patients who are bleeding is the assessment of whether platelets are more reactive than they should be. This question can arise in the setting of unexplained arterial thrombosis or when the impact of antiplatelet agents is being evaluated. Management of antiplatelet agents is discussed in the last part of this section. The approaches used to evaluate increased platelet function tend to be relatively nonspecific. The classic marker of platelet hyperactivity is “spontaneous” aggregation observed in the platelet aggregometer, generally defined as occurring when platelet-rich plasma is stirred at 37°C without adding an agonist. As with aggregometry, in general, observations of spontaneous aggregation are only reliable if the blood has been obtained, with care taken to avoid generation of ADP or thrombin. A related test is to look for a leftward shift in the dose-response curves for multiple platelet agonists so that platelet aggregation consistently occurs at lower than normal agonist concentrations. This method requires careful establishment of normal ranges in a large cohort of healthy individuals studied in the same laboratory. Evidence for platelet activation occurring in vivo can obtained by measuring molecules that are secreted by activated platelets or that are shed from the surface of activated platelets. Sensitive ELISA (enzyme-linked immunosorbent assay)-based assays are available for the α-granule proteins, platelet factor 4, and β-thromboglobulin, and for proteolytically shed P-selectin, GP Ibα (glycocalicin), and CD40L. Each of these assays has its own strengths and weaknesses.32 Evidence for platelet activation in vivo can also be obtained by using flow cytometry to examine platelets ex vivo for evidence of degranulation or allbb3 activation. The former is often detected with antibodies to P-selectin; the latter with a conformation-specific antibody such as PAC-1.
Identifying Possible Mechanisms.
Platelet function defects can be hereditary or acquired. The hereditary defects have proven to be enormously valuable for the insights that they provide into normal platelet biology, which, in turn, has led to ways to help the patients who have them.5 Hereditary defects are, however, rare. Acquired defects arise through the use of medications, such as aspirin, the presence of diseases that impact platelet function (eg, diabetes, uremia, and myeloproliferative disorders), and the presence of antibodies that can impair platelet activation (eg, idiopathic thrombocytopenic purpura). Acquired defects are more common than inherited disorders, but often less clearly defined in terms of the mechanism. Some of the better-characterized hereditary defects can be at least tentatively diagnosed through platelet function testing, particularly in the aggregometer.24,25,33,34 Glanzmann's thrombasthenia, for example, results in platelets that will undergo shape change, but not form aggregates because of the absence of functional αIIbβ3. This produces essentially flat aggregation traces with each of the agonists that are typically used in clinical testing (ADP, collagen, epinephrine, a thrombin receptor agonist peptide [eg, SFLLRN], and a TxA2 analog [eg, U46619]), but not with ristocetin, which causes platelet agglutination by the VWF. The presence of normal amounts of αIIbβ3 on the platelet surface can be detected in a flow cytometer using any of several monoclonal antibodies that recognize one or both of its subunits. The ability of αIIbβ3 to be activated can be detected with PAC-1, an antibody that recognizes only the active conformation of αIIbβ335 or fluorescently tagged fibrinogen.
Other well-characterized monogenic platelet defects include the Bernard-Soulier syndrome, myosin (MYH9) defects, a variety of storage pool defects that affect secretion, and very rare defects that affect collagen receptors (GP VI and α2β1 integrin). These disorders vary in their clinical impact, but collectively represent examples of when platelet function testing can be predictive of clinical effects. Diagnosis begins with personal and family histories, paying close attention to medications (prescribed and over the counter), and a physical examination. If the history and physical suggest that a definable problem is present, they are followed by lumi-aggregometry to assess secretion, as well as aggregation, additional testing to narrow down the precise defect, and, when appropriate, genetic testing.36 Dense granule secretion defects can be identified by looking for the absence of a secretion-dependent “second wave” of platelet aggregation and then further evaluated using the lumi-aggregometer to detect ATP release. Dense granule formation can be assessed by measuring platelet ADP:ATP ratios in resting platelets. α-Granule release is detected by using flow cytometry to identify P-selectin on the surface of activated platelets. The formation and filling of platelet secretory granules can also be studied with transmission electron microscopy.
Hereditary defects are not limited to well-characterized monogenic defects that affect essential platelet proteins. As with all other cells and systems in humans, platelet function is affected by the collective impact of polymorphisms that alter protein expression and function. Such differences might, for example, account in part for reproducible variations among individuals in responses to specific agonists or drugs.37–39 This is still a research question and not one that is yet amenable to evaluation in the clinical laboratory or, for that matter, an easy translation into predictable clinical impact. Finally, a short list of mutations in transcription factors, such as GATA-1 and RUNX-1, can impact platelet function, presumably by affecting the production of proteins from multiple genes that are normally expressed by megakaryocytes and then placed into platelets. These are a different type of “polygenic” defect.
Acquired defects are even more heterogeneous. Some, such as the effects of aspirin on TxA2 production, are very well characterized. Lumi-aggregometry in this case typically shows a secretion defect that can be overcome by raising the agonist concentration and a specific defect in platelet responses to exogenous arachidonate that can be bypassed by substituting a TxA2 analog, such as U46619. The clinical impact of aspirin is also well understood (minimal unless another defect such as von Willebrand's disease is also present). On the other hand, systemic disorders, such as uremia, can have pleiotropic effects on platelet function tests with variable effects on hemostasis in the clinical setting that are not necessarily predicted by nor correlative with the results of laboratory testing. In other words, defects in the clinical laboratory most reliably translate into problems in the patient when those defects are well characterized and augmented by a body of clinical experience.
As is frequently done in the context of deep venous thrombosis, it is worth considering how many members of a large group of patients with clinical histories suggestive of a platelet function defect will prove to have something that is well understood by present technology. In recent reviews of clinical experience at specialty centers, the fraction of patients with a specific diagnosis at the end of the workup was less than 50% and even lower if VWF deficiency is excluded.40,41
Assessing the Efficacy of Antiplatelet Agents.
The widespread use of antiplatelet agents to prevent cardiovascular events has inevitably led to attempts to use platelet function testing to adjust dosing and determine the etiology of “drug resistance.” If, for example, a patient on aspirin has a second myocardial infarct, that is because he/she has not taken the drug, TxA2 formation has not been fully suppressed at the dose prescribed, or the extent of vascular disease overwhelms the impact on platelet activation of abolishing TxA2 production. Similar questions arise in patients receiving P2Y12 antagonists: is the failure of the drug to prevent a second event (or stent occlusion) because of noncompliance, inadequate dosing, polymorphisms affecting conversion of the drug to its active metabolite, or an overwhelming impetus for platelet activation that cannot be suppressed by a receptor blockade?
A number of devices have now been introduced and, in at least one case, approved for helping to manage patients on antiplatelet agents (Table 2). As a concept, the idea is extremely attractive. In practice, the benefits have been more elusive. What has been shown is that aspirin resistance in healthy individuals is rarely, if ever, due to a failure to inhibit TxA2 formation42 and that patients with recurrent cardiovascular events while on P2Y12 antagonists are more likely to have laboratory evidence of less blockade of their ADP receptors.43–45 This does not mean, however, that the converse is necessarily true; that is, that efficacy (or the lack of efficacy) can be necessarily be predicted from a laboratory test and that dosing of the drug should be adjusted accordingly.
Summary: How Useful Are Platelet Function Tests in the Clinical Setting?
Platelet function tests may be far less expensive than a series of magnetic resonance imaging scans, but they are neither inexpensive nor undemanding of technician time. Therefore, in closing, it is worth considering whether they really are useful in the clinical setting. The answer is a solid “it depends.” Platelet function testing can be uniquely useful for establishing the presence of well-characterized hereditary and acquired defects of platelet function, but as already noted, there are many patients with a reasonable history of mucocutaneous bleeding suggestive of a platelet defect in whom no molecular diagnosis can yet be established, even when abnormalities have been documented in standard platelet function tests. All too often, the workup shows a pattern of abnormal aggregation traces that is neither diagnostic of a specific defect nor predictive of clinical impact. A good example is an “aspirin-like defect” in a patient not on aspirin or any other COX-1 inhibitor. Similarly, at the other end of the clinical spectrum, aggregation studies and flow cytometry markers (eg, surface P-selectin expression and PAC-1 binding) may document platelet overreactivity, particularly in the setting of vascular disease or dyslipidemia, but not provide further insight into thrombosis risk in that particular patient. Keeping in mind that people rarely undergo comprehensive platelet function in the absence of a suggestive history, the predictive value of the workup is then far more likely to be based on the clinical history far than the test results. Assessing the impact of antiplatelet agents is clearly an important priority, given the prevalence of cardiovascular disease and the enormous quantities of antiplatelet agents that are prescribed every year. Although there is still no widely accepted equivalent of the international normalized ratio for adjusting the dosage of antiplatelet agents, some progress has been made for specific drugs in particular settings, such as P2Y12 antagonists and stent thrombosis. Better laboratory tests that reflect platelet function in vivo and more experience with these tests in clinical trials are clearly indicated, particularly if a goal is to close the loop between laboratory testing and clinical decision making. Until that happens, hematologists will have to continue to rely on their clinical judgment plus an array of useful, but imperfect, assessment tools.46
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests.
Off-label drugs: None disclosed.
Correspondence
Lawrence Brass, MD, PhD, Hematology-Oncology Division, Department of Medicine, and the Department of Pharmacology, University of Pennsylvania School of Medicine, 421 Curie Blvd., Room 915 BRB-II, Philadelphia, PA 19004; Phone: (215) 573-4669; e-mail: Brass@mail.med.upenn.edu
