
Abstract
Modern treatment approaches such as chemoimmunotherapy (e.g., fludarabine/cyclophosphamide/rituximab or FCR) are highly effective in the majority of chronic lymphocytic leukemia (CLL) patients. However, there remains a small but challenging subgroup of patients who show ultra high-risk genetics (17p deletion, TP53 mutation) and/or poor response to chemoimmunotherapy. The median life expectancy of these patients is below 2 to 3 years with standard regimens. Accordingly, CLL with the 17p deletion (and likely also with sole TP53 mutation) should be treated with alternative strategies. While p53 defects appear to play a central role in our understanding of this ultra high-risk group, at least half of the cases will not be predictable based on existing prognostic models. Current treatment approaches for patients with p53 defects or poor response to chemoimmunotherapy should rely on agents acting independently of p53, such as alemtuzumab, lenalidomide, flavopiridol, and a growing number of novel compounds (or combinations thereof) currently available in clinical trials. Poor survival times of patients with ultra high-risk CLL suggest that eligible patients should be offered consolidation with reduced-intensity allogeneic stem-cell transplantation or experimental approaches in clinical trials.
Introduction
In contrast to most other types of leukemia, chronic lymphocytic leukemia (CLL) is not treated upon diagnosis, but upon progression to symptomatic disease.1 This approach is based on the results of randomized trials failing to show benefits of unstratified early treatment with chemotherapy. However, over the last decades of CLL treatment, we have seen the transition from single-agent, alkylator-based therapies to nucleoside analogs, combination chemotherapy, and, most recently, chemoimmunotherapy (e.g., combining monoclonal antibodies such as rituximab with chemotherapy such as fludarabine + cyclophosphamide, which is called FCR).2 As a result, complete response rates have improved from 3% to 7% with chlorambucil to a maximum of 70% with FCR.3–7 In the randomized German CLL Study Group (GCLLSG) CLL8 trial comparing FC and FCR, the complete response rate in the FCR arm was 44%.5 About 90% to 95% of patients receiving FCR as a first-line treatment are expected to respond. Historical comparisons suggest that for the first time, overall survival (OS) has been improved with the application of chemoimmunotherapy.8,9 In addition, recent results suggest that modern therapies are changing the natural course of CLL: multivariate analysis including genetics (FISH, TP53 mutation, IGHV) within prospective trials (GCLLSG CLL4: F vs. FC trial), and, most recently, results of the CLL8 (FC vs. FCR) trial suggest that the initial treatment choices influence OS. However, in all available analyses, there is a strong association between genetic disease profile and outcome.10 Therefore, the development of prognostic and predictive models leading to stratified treatment strategies for specific disease subgroups are of importance. While most patients with CLL will either not need treatment or will respond well to treatment, there remains a group of patients with refractory CLL, as defined by the International Workshop on Chronic Lymphocytic Leukemia (IWCLL) and the National Cancer Institute (NCI) working groups,11 ) or suboptimal response (early relapse after exposure to FCR or regimens of similar potency). Identifying this group of patients early, ideally before treatment, by clinical or biological parameters and designing adequate treatment options remain key issues in the treatment of CLL.12 This will benefit not only the ultra high-risk group but other patients as well, because targeted approaches can also be curtailed to define low-risk groups. For this review, we have defined ultra high-risk CLL as patient subgroups with median survival below 24 to 36 months at the treatment time point. Based on this definition, patients with the 17p deletion, the TP53 mutation, and F-refractory CLL belong to this group, as well as patients with suboptimal response to intense treatment (e.g., FCR) (Figure 1).

Definition of ultra high-risk CLL and incidence of the specific subgroups. The definitions for the purpose of this article are given with regard to biological (i.e., 17p deletion, TP53 mutation) and clinical (i.e., response to treatment) characteristics.
Definition of ultra high-risk CLL and incidence of the specific subgroups. The definitions for the purpose of this article are given with regard to biological (i.e., 17p deletion, TP53 mutation) and clinical (i.e., response to treatment) characteristics.
It is important to stress that other prognostic factors associated with inferior outcome in CLL, such as the 11q deletion, unmutated IGHV, ZAP70 expression, or ß2-microglobulin levels, will not be discussed here. While these are clearly associated with poor prognosis, they cannot be included in the ultra high-risk category of patients treated with chemoimmunotherapy according to the definition given above.5,13
Understanding Ultra High-Risk CLL
Molecular Mechanisms Underlying Refractory CLL
p53 plays a central role in our current understanding of why CLL patients fail to respond to chemo(immuno)therapy and have short survival times. The single best-characterized molecular lesions found in refractory CLL are the deletion 17p13 (TP53 locus) and TP53 mutations.6,14–17
Poor outcome of patients with the 17p deletion has been observed for over 15 years, and the initial single-center experience documenting the failure to induce remissions with purine analog-based chemotherapy in this group of patients18 has been confirmed in all prospective trials since.3,14,19 Indeed, this group of patients (5%–8% of patients receiving first-line treatment)13,14,19,20 are the first group to be treated in separate (genotype-specific) treatment trials in CLL. This is based on the observation that median OS will be in the range of 2 to 3 years from the time of front-line treatment. While there is a small subgroup of patients with the 17p deletion (and mostly mutated IGHV) without treatment indications who may exhibit stable disease for years,21,22 the outcome of most patients with the 17p deletion and need for treatment is very poor.13,14,19,20 Patients with the 17p deletion will very rarely achieve complete response after chemoimmunotherapy, as demonstrated in the CLL8 trial, in which the group without the 17p deletion had a complete response rate of 21.8% (FC) or 44.1% (FCR) compared with 2.3% among patients with the 17p deletion (both arms).5
Over 80% of CLL patients with the 17p deletion also carry a TP53 mutation on the remaining allele.23–25 There have been a number of retrospective studies suggesting that, in addition to the 17p deletion, the TP53 mutation even in the absence of the 17p deletion predicts poor outcome.24–27 In a recent analysis of the GCLLSG CLL4 trial (F vs. FC), we found TP53 mutations in 8.5% of patients (28 of 328; 4.5% in the absence of the 17p deletion).23 No complete response was observed in patients with the TP53 mutation. Median progression-free survival (PFS) (23.3 vs. 62.2 months) and OS (29.2 vs. 84.6 months) were significantly decreased in the group with the TP53 mutation (both p < 0.001). PFS and OS for patients with the 17p deletion and patients with the TP53 mutation in the absence of the 17p deletion were similar (Figure 2). Multivariate analysis identified the TP53 mutation as the strongest prognostic marker regarding OS (hazard ratio [HR] 7.2, p < 0.001). Other independent predictors of OS were unmutated IGHV status (HR 1.9), 11q deletion (HR 1.9), 17p deletion (HR 2.3), and FC treatment (HR 0.6).

CLL with TP53 mutation without the 17p deletion shows similar outcome as CLL with the 17p deletion after first-line F-based therapy (CLL4 trial of the GCLLSG). OS of the group with the 17p deletion (n = 16) is shown in red; OS of the sole TP53 mutation without the 17p deletion (n = 14) is shown in black; and the OS of remaining patients (n = 277) is shown in green. Median OS was significantly shorter for patients with the 17p deletion (19.2 months) and sole TP53 mutation (30.2 months) compared with patients without either abnormality (median not reached; p < 0.001). (Data from 23 .)
CLL with TP53 mutation without the 17p deletion shows similar outcome as CLL with the 17p deletion after first-line F-based therapy (CLL4 trial of the GCLLSG). OS of the group with the 17p deletion (n = 16) is shown in red; OS of the sole TP53 mutation without the 17p deletion (n = 14) is shown in black; and the OS of remaining patients (n = 277) is shown in green. Median OS was significantly shorter for patients with the 17p deletion (19.2 months) and sole TP53 mutation (30.2 months) compared with patients without either abnormality (median not reached; p < 0.001). (Data from 23 .)
Early results from the CLL8 trial suggest that patients with the TP53 mutation only, the 17p deletion without the TP53 mutation, and both the mutation and the deletion have poor response to FC and FCR.28 In contrast, data from the ECOG 2997 trial and the UK LRF CLL4 trial did not show such profound effects of the TP53 mutation in the absence of the 17p deletion, but technical approaches and incidence of mutations and the choice of treatment (UK LRF4) were different.14,29 In general, it appears that the impact of biological prognostic markers such as the TP53 mutation and the 17p deletion is the most pronounced when more potent treatments (i.e., FC or FCR) are applied, likely due to the fact that the outcome of most subgroups is markedly improved, while the outcome of the ultra high-risk patients remains essentially unchanged. Further evidence for the resistance of clones with the 17p deletion and the TP53 mutation comes from the observation that these aberrations are the most common abnormalities acquired during the course of the disease (clonal evolution), and, importantly, expand under the selection pressure of chemo(immuno)therapy.24,30,31 With advanced treatment lines, the incidence of TP53 aberrations increases from approximately 10% (first-line), to approximately 20% at relapse and as much as 40% to 50% in F-refractory CLL.10,17
Because CLL with the TP53 mutation carries a very poor prognosis regardless of the presence of the 17p deletion when treated with F-based chemotherapy, TP53 mutation analysis should be incorporated into the evaluation of CLL patients before treatment initiation. CLL patients with the TP53 mutation and indication for therapy should be considered for alternative treatment approaches up front because they are at ultra high risk. These findings are of particular importance because the group of patients who should be considered for alternative approaches (genotype-specific) will increase from 5% to 8% up to 8% to 12% for front-line therapy when considering the TP53 mutation in addition to the 17p deletion.23,28
Even in spite of the strong association between poor clinical outcome and the TP53 mutation, p53 abnormalities will only explain a fraction of refractory CLL cases. The proportion of F-refractory CLL cases carrying the TP53 loss or mutation has been estimated to add up to 40 to 50.16,17 Current data suggest that 29% to 52% of F-refractory CLL cases (treated with F or FC/FCR) in first-line-treatment settings may be explained by the TP53 mutation and/or the 17p deletion.23,28
These findings raise questions regarding the basis of refractoriness in the remaining cases. While there are numerous studies showing defects in the DNA-damage machinery in CLL, this issue has not been adequately addressed in larger studies. There is some evidence that additional cases are associated with p53 pathway defects such as miR-34a dysregulation,32 but the mechanisms of refractory CLL remain an important research question that is best addressed by diverse biological analyses of carefully annotated samples from clinical trials.
Clinical Definition of Ultra High-Risk CLL Based on Response
While it is important to identify new prognostic and predictive factors, there is a clear picture emerging that, even in the absence of known ultra high-risk markers, poor response to intensive therapy (e.g., FCR) will be associated with short OS and can be considered ultra high risk. Results from the M.D. Anderson Cancer Center (MDACC)8,33 (Keating et al., personal communication) and from the CLL8 trial (FC vs. FCR) suggest that OS is very poor for patients who need early (within 2–3 years) re-treatment after first-line therapy.
In the pivotal trial cohort from MDACC establishing FCR as the treatment with the highest efficacy in CLL,4,8 patients with a PFS below 36 months had a significantly shorter OS (<24 months from subsequent treatment) compared with patients with a PFS > 36 months (p < 0.01) (Keating et al., personal communication). While this was a single-institution trial, very similar results are becoming available from randomized multi-center trials. In an analysis from the GCLLSG CLL8 trial (FC and FCR arm), patients with a PFS below 12 months (n = 47), 12 to 24 months (n = 45), or above 24 months showed significant differences in OS from the time point of second-line treatment (median 13.1, 20.3, and 44.6 months, respectively; p < 0.01) (Figure 3). These data suggest that patients with PFS below 24 months should be considered ultra high risk at the time of recurring treatment indication. It is important to note that similar outcome can be expected in patients treated with comparable regimens incorporating other chemotherapy agents such as pentostatin, mitoxantrone, and bendamustine (e.g. PCR, FCM-R, BR).

Patients relapsing early after FC or FCR have a poor outcome. OS from the time point of second-line treatment for patients initially treated in the CLL8 trial (FC vs. FCR) according to remission duration after first-line treatment. Patients with short remissions (<24 months) have very poor outcomes, suggesting that these patients should be considered for investigational treatment and allo-SCT.
Patients relapsing early after FC or FCR have a poor outcome. OS from the time point of second-line treatment for patients initially treated in the CLL8 trial (FC vs. FCR) according to remission duration after first-line treatment. Patients with short remissions (<24 months) have very poor outcomes, suggesting that these patients should be considered for investigational treatment and allo-SCT.
While these observations will only be relevant to a small subgroup of patients (about 10% of patients treated in first-line; e.g., 92 of 814 [11.3%] in the CLL8 trial), it is an important group of patients. In the design of trials, this group should be addressed separately from late relapses or relapses after non-intense pretreatment. In many ways, the classical definition of F-refractory CLL (no remission or remission under 6 months in duration1 ) and corresponding risk evaluation is currently undergoing reassessment in the era of chemoimmunotherapy.
Treatment of Ultra High-Risk CLL
Targeted Treatment/Genotype-Specific Treatment in CLL
Ideally, the treatment approach should target the molecular lesion critical in ultra high-risk CLL. This can be envisioned in cases of CLL with mutant TP53. Remarkable progress has been made in the development of p53-binding molecules that can rescue the function of certain p53 mutants. Agents have been identified that exploit the p53 pathway by either seeking targets and compounds that show synthetic lethality with TP53 mutations or by looking for non-genotoxic activators of the p53 response.34 While there is some evidence that cells with mutant TP53 may exhibit specific vulnerabilities, clinical translation of this is so far inconclusive.34–36 The approach currently taken for CLL with loss or mutation of TP53 and F-refractory CLL relies mostly on the use of agents acting independently of the DNA damage pathway.37 While the success of this approach has been documented for antibodies (e.g., alemtuzumab), there is still considerable room for improvement. Most current treatment recommendations or approvals from regulatory authorities are based on non-randomized trials.38,39 There is little evidence that patients with the 17p deletion/TP53 mutation or FCR-refractory CLL will benefit from alternative chemotherapy regimens, because clinical trials show similar outcomes for these patients after treatment such as BR or R-FCM.40,41
As shown in Table 1, most regimes used in the setting of CLL with the 17p deletion or F-refractoriness can be expected to induce responses in 30% to 50% of patients with relatively short PFS. Based on these results and the data presented above, these patient groups are prime candidates for novel clinical trials (Figure 4). Ideally, these should be randomized or multi-arm studies using novel agents for which there is at least some evidence for specific activity in ultra high-risk CLL (Table 1).
ORR and PFS for patients with the 17p deletion or F-refractory CLL after different treatment approaches
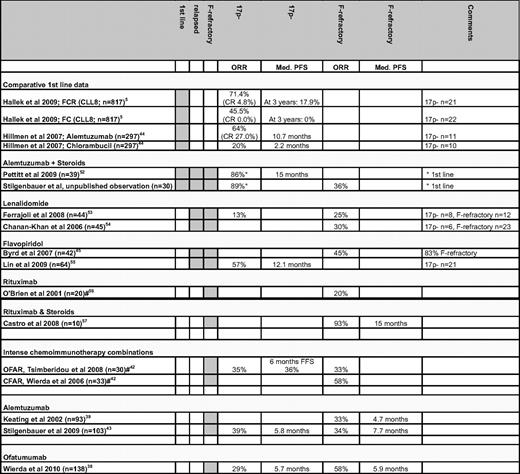
#Data shown are only for patients with F-refractory CLL.

Suggested definitions and corresponding treatment algorithms for patients with ultra high-risk CLL. Three groups are defined: 1) patients with the 17p deletion or the TP53 mutation and treatment indication, 2) patients with F-refractory CLL, and 3) patients with short PFS after intense therapy (e.g. FCR). Treatment results with current “standard” chemo(immuno)therapy are poor, and therefore these patients are prime candidates for clinical trials evaluating novel agents (that would ideally be comparative). The choice of agent/trial should be based on evidence for efficacy available in the specific situation (i.e., the TP53 mutation). In addition, these patients are prime candidates for consolidation with reduced intensity allo-SCT (fit patients) or experimental approaches in clinical trials. These suggestions are rarely based on comparative trial data because these are scarce.
Suggested definitions and corresponding treatment algorithms for patients with ultra high-risk CLL. Three groups are defined: 1) patients with the 17p deletion or the TP53 mutation and treatment indication, 2) patients with F-refractory CLL, and 3) patients with short PFS after intense therapy (e.g. FCR). Treatment results with current “standard” chemo(immuno)therapy are poor, and therefore these patients are prime candidates for clinical trials evaluating novel agents (that would ideally be comparative). The choice of agent/trial should be based on evidence for efficacy available in the specific situation (i.e., the TP53 mutation). In addition, these patients are prime candidates for consolidation with reduced intensity allo-SCT (fit patients) or experimental approaches in clinical trials. These suggestions are rarely based on comparative trial data because these are scarce.
There are numerous targeted strategies aiming at key pathways (e.g. PI3-kinase, NFkB, SYK, AKT) and a number of antibody-based approaches targeting alternative epitopes (e.g., CD19, CD20, CD23, CD37) currently available in clinical trials. A detailed discussion of these is beyond the scope of this article. Figure 5 gives an overview of a selection of substances in clinical development in CLL.

Selection of targets and novel agents in early clinical trials of CLL. There are multiple “biological” therapeutic approaches in current clinical development in CLL. Signal transduction inhibition will often target the B-cell receptor cascade, which has been shown to be critical to CLL cell survival. A number of surface molecules of CLL cells can be targeted by antibodies. Different members of the apoptosis machinery are targets currently explored in clinical trials. Microenvironmental stimulation and T-cell interaction are of critical importance for CLL cell survival, and are being increasingly utilized for treatment strategies. There is ongoing interest in using mutant p53 as a “druggable” target, and a number of agents have been identified that may preferentially target mutant p53.34 Improving efficacy in CLL with p53 pathway defects is likely to offer the greatest overall benefit.
Selection of targets and novel agents in early clinical trials of CLL. There are multiple “biological” therapeutic approaches in current clinical development in CLL. Signal transduction inhibition will often target the B-cell receptor cascade, which has been shown to be critical to CLL cell survival. A number of surface molecules of CLL cells can be targeted by antibodies. Different members of the apoptosis machinery are targets currently explored in clinical trials. Microenvironmental stimulation and T-cell interaction are of critical importance for CLL cell survival, and are being increasingly utilized for treatment strategies. There is ongoing interest in using mutant p53 as a “druggable” target, and a number of agents have been identified that may preferentially target mutant p53.34 Improving efficacy in CLL with p53 pathway defects is likely to offer the greatest overall benefit.
Treatment of Patients with F-Refractory CLL
Patients with F-refractory CLL, today mostly referring to F-based chemo(immuno)therapy combinations, have a very poor prognosis.42 Response to various chemotherapeutic approaches is low in this setting, and alternative approaches (acting independently of the DNA damage pathway) are preferred (Figures 4 and 5). The anti-CD52 antibody alemtuzumab is well studied in patients with refractory CLL, and is documented to induce remission independent of the genetic make-up of the CLL cells.43,44 Responses can be expected in about one-third of patients with F-refractory CLL and in 60% to 80% of treatment-naive patients; however, remission duration is short if no consolidation strategies are applied.
There is relatively little information on the use of rituximab in F-refractory CLL, but it is used in combination with a number of agents this setting (e.g., steroids). Recently, the novel CD20 antibody ofatumumab has been assessed in a phase II study of patients with F- or F + alemtuzumab-refractory CLL. Results showed an encouraging 50% overall response rate (ORR; assessed during ongoing treatment), but a relatively short PFS of 5.9 months.38 Activity in CLL with the 17p deletion has not been clearly demonstrated.
Flavopiridol is a broad, cyclin-dependent kinase inhibitor that induces apoptosis in leukemic cell lines and CLL cells, and its activity is p53 independent. In the pivotal study by Byrd et al., 45% of patients with high-risk CLL responded, and their PFS was 12 months.45 Currently, flavopiridol is being evaluated in an international multicenter phase II study of F-refractory CLL to further explore its use in this setting.
Lenalidomide has been used in advanced CLL. Much of the agent's appeal is based on a mechanism of action that is, even if incompletely understood, very different from chemotherapy or other agents.46 Lenalidomide has been studied in a limited number of patients with the 17p deletion or F-refractory disease, and the results are summarized in Table 1. Current approaches with this agent explore combinations and maintenance/consolidation strategies.
Treatment of Patients with Suboptimal Response to Intensive Chemoimmunotherapy
Similarly to OS, which correlates well with remission duration after FCR, the ORR after second-line therapy will depend on the length of initial remission. In an analysis of the response to subsequent treatment of patients in the initial FCR trial,4,8 the ORR to intense second-line treatment was 44% (PFS after FCR < 36 months) compared with 70% (>36 months) (Keating et al., personal communication). The clinical consequences of this observation should influence the choice of relapse treatment and consolidation approaches (e.g., allogeneic stem-cell transplantation [allo-SCT]). Based on results from MDACC, a potential approach to these patients (PFS < 24–36 months after initial FCR) would be to consider re-treatment with FCR or similar regimens in patients who responded to first-line treatment with a complete response or nodular partial response, because the ORR is 68% to 78% in these patients. On the other hand, patients who only had a partial response after first-line FCR treatment should be treated with investigational agents, because responses will be in the range of only 30% (M. Keating et al., personal communication) (Figure 4). The results of CLL8 confirm that patients with short response durations (<24 months) after initial treatment with FC or FCR should be considered candidates for trials investigating alternative approaches (Figures 3, 4, and 5).
Currently, evidence is accumulating from prospective trials that a refined guidance of treatment will be possible by assessing minimal residual disease (MRD). A number of studies have shown the association of improved outcome with MRD negativity.47,48 Correspondingly, high MRD levels correlate with poor outcome after chemo(immuno)therapy, and it is foreseeable that MRD assessment could allow further risk stratification early during or after treatment to identify high-risk or ultra high-risk patients.49
Allo-SCT
The recent past has seen a surge in transplantations for patients with CLL. This has been supported by the use of reduced-intensity conditioning regimens, improved (genetic) risk stratification, and the availability of more data on outcome after allo-SCT. A recent proposal by international experts has defined a consensus on when to consider allo-SCT in CLL.50 Although “poor-risk CLL” had not been defined unequivocally at that time, allo-SCT was proposed for younger patients who do not respond to or relapse early (< 12 months) after therapy with purine analog, those who relapse within 24 months after purine analog-based combination therapy or autologous transplantation, and those with TP53 abnormalities (deletions or mutations) requiring treatment. Indeed, based on the recent data described above, this recommendation can be fully endorsed.
Clinical trial data suggest that the 4-year OS of high-risk CLL patients receiving a reduced-intensity allo-SCT is between 48% and 70%.51 In the setting of allo-SCT, the ultra high-risk features 17p deletion or F-refractoriness appear not to affect efficacy per se. In contrast, disease status at the time of SCT (i.e., lack of remission or bulky lymphadenopathy) strongly predicts failure. This again underlines the need for alternative approaches for remission induction in ultra high-risk CLL patients to make reduced-intensity allo-SCT a feasible option. Prospective trials are warranted to define the optimal transplant strategies best suited for different clinical scenarios. In addition, there is an urgent need to explore non-transplant consolidation options, because the majority of ultra high-risk CLL patients will not be transplant candidates due to their age and comorbidities.
Summary
We currently consider patients with the 17p deletion or TP53 mutation who need treatment the only established ultra high-risk CLL group based on biological markers. However, there is growing evidence that the group of patients with F-refractory disease or early relapse (<24–36 months) after FCR or similar treatment should also be considered ultra high risk. Ultra high-risk patients should be considered for treatment trials with novel compounds and combinations, and they are prime candidates for allo-SCT as consolidation. Further investigation of other biological causes in addition to p53 defects is of prime importance not only to enhance our understanding of ultra high-risk CLL, but also to help in its management.
Acknowledgments
We are indebted to Michael Keating for sharing unpublished results and helpful discussions vital to the manuscript. We thank Asher Chanan-Khan for sharing unpublished data and Peter Dreger, Andreas Viardot, Michael Hallek, Alessandra Ferrajoli, John Byrd, and Michael Keating for critical reading of the manuscript. We wish to acknowledge the important contributions of the numerous investigators whose work could not be cited due to space restrictions. We acknowledge the GCLLSG (chairman Michael Hallek) and the European Research Initiative on CLL (ERIC, chairman E. Montserrat) for support and discussions. We thank Hartmut Döhner for longstanding support and mentoring.
Disclosures
Conflict-of-interest disclosure: SS has consulted for and received research funding and honoraria from Roche, Mundipharma, GSK, Genzyme, Boehringer-Ingelheim, Bayer, Amgen, and Sanofi Aventis. TZ has been a member of advisory boards and has received honoraria from Boehringer-Ingelheim, Celgene, GSK, Trubion, and Bayer-Schering Pharma. Off-label drug use: Off-label use of diagnostic tests and therapeutic agents.
Funded in part by the DFG (DFG STI 296/2–1), German José Carreras Leukemia-Foundation (R06/28v, R08/26f), Else Kröner-Fresenius-Stiftung (P20/07//A11/07), Deutsche Krebshilfe (108355, 106142), and the Global CLL Research Foundation.
Correspondence
Dr. Stephan Stilgenbauer, Department of Internal Medicine III, University of Ulm, Albert-Einstein-Allee 23, 89081 Ulm, Germany; Phone: 49–731-500–45521; Fax: 49–731-500–45525; e-mail: stephan.stilgenbauer@uniklinik-ulm.de
