
Abstract
The 21st century ushered in the dawn of a new era of targeted therapeutics and a dramatic shift in the management of chronic-phase chronic myeloid leukemia (CP-CML) patients. Groundbreaking scientific and translational studies have led to the rapid development and approval of several effective BCR-ABL tyrosine kinase inhibitors (TKIs). In the United States, there are currently 3 approved BCR-ABL TKIs for newly diagnosed CP-CML patients. It is anticipated that clinical outcomes will continue to improve as more TKIs that address unmet medical needs are approved. However, to achieve this goal, it is critical to carefully monitor and optimally manage patients. To this end, the latest seminal clinical trial results of approved and investigational BCR-ABL TKIs and some of the salient unique features of each of these agents are summarized herein.
Introduction
In the last decade, the development of effective tyrosine kinase inhibitors (TKIs) for patients with chronic-phase chronic myeloid leukemia (CP-CML) has revolutionized the treatment of a disease that hitherto had only allogenic stem cell transplantation as a treatment modality capable of reliably imparting long-term disease control. Since the initial US Food and Drug Administration (FDA) approval of imatinib in 2001, treatment milestones have been reached established based upon their impact on subsequent disease progression and overall survival (OS), and an appreciation of resistance mechanisms has led to the development of more effective and potent BCR/ABL inhibitors. As a result, CP-CML patients diagnosed today who are capable of adhering to effective oral TKI therapy on a chronic basis are expected to have an excellent probability for achieving long-term survival, and perhaps even a normal life expectancy, with preservation of an acceptable quality of life.
Clinical response, depth of response, and the impact on disease outcome
Clinical response to imatinib can be defined with increasing depth by hematologic, cytogenetic, and molecular criteria. Complete hematologic response has been defined as a reduction in WBCs to < 10 000/mm3. Cytogenetic response has been classically defined based upon evaluation of a minimum of 20 bone marrow (BM) metaphases, partial cytogenetic response is defined as 1%-35% Philadelphia chromosome–positive (Ph+) BM metaphases, and complete cytogenetic response (CCyR) as 0% Ph+ metaphases. Molecular response is determined by quantifying the BCR-ABL transcript level through real-time quantitative PCR. Longer-term follow-up of patients enrolled on the International Randomized Study of Interferon versus STI571 (IRIS) has made it clear that the depth of initial cytogenetic response to imatinib is highly predictive of risk for disease progression and OS. In the 5-year analysis of the IRIS data, a CCyR at 12 months was associated with freedom from progression to accelerated phase (AP) or blast crisis (BC) in 97% of patients at 60 months follow-up compared with 81% of patients without a major cytogenetic response (0%-35% Ph+ metaphases) (P < .001).1 In a large single-institution analysis of 204 consecutive newly diagnosed CP-CML patients treated with imatinib, CCyR at 12 months was associated with better OS 98.0% versus 74.1% (P = .03) and progression-free survival (PFS) 96.0% versus 74.0% (P = .007), respectively, compared with those not achieving CCyR.2 Moreover, patients with a partial cytogenic response at 12 months (1%-35% Ph+ metaphases) also had poorer PFS (82%, P = .01). The minimally acceptable level of response on imatinib at present is CCyR, achieved no later than 12-18 months.
The contribution of molecular response to PFS on first-line imatinib therapy has been analyzed. In the IRIS study, patients who achieved a major molecular response (MMR; defined as a reduction in levels of BCR-ABL transcript to ≤ 0.1% of the average level of BCR-ABL transcript in 30 untreated CP-CML patients) by 18 months had 100% freedom from progression to AP/BC and 95% event-free survival (EFS; “events” defined as death from any cause, progression to AP/BC, loss of a complete hematologic response or major cytogenetic response, or an increasing white cell count to > 20 × 109/L) at 7 years with only a 3% probability of loss of CCyR compared with 26% for patients with CCyR but not MMR (P < .001).3 In addition, patients with BCR-ABL transcripts > 10% at 6 months and > 1% at 12 months had inferior EFS and higher rate of progression to AP/BC compared with all other molecular response groups.
The degree to which achievement of MMR adds additional clinical benefit to the achievement of CCyR is presently unclear. As stated above, for CP-CML patients with TKIs, deeper cytogenetic responses have convincingly translated to better long-term outcomes. Evolving data therefore suggest that faster and deeper molecular responses portend a more favorable prognosis. Therefore, aiming to achieve MMR may be justifiable, particularly in younger CP-CML patients in whom the goal of treatment is the preservation of a normal lifespan. However, it must be stated that with 8 years of follow-up of patients treated with imatinib, the added clinical benefit of MMR appears to be incremental over the benefit imparted by CCyR. The true benefit of MMR achievement will likely be more precisely defined with longer follow-up times.
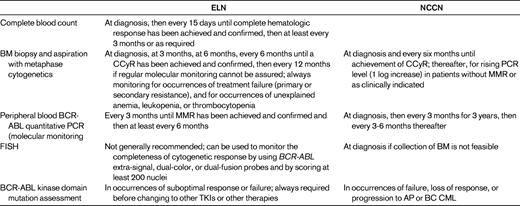
Efforts to distinguish patients who are likely to do well on imatinib from those whose outcome is not as likely to be favorable have led the European LeukemiaNet (ELN)4 and National Comprehensive Cancer Network (NCCN)5 to develop monitoring recommendations (Table 1). Cytogenetic response based upon BM metaphase analysis has been extensively studied and correlated with outcomes on medical therapies for CP-CML and is therefore considered the “gold standard” for monitoring disease response. Whereas FISH offers the convenience of assessing the peripheral blood, its sensitivity is not significantly better than metaphase karyotype analysis and it has not been prospectively validated as a monitoring tool in clinical studies. Real-time quantitative BCR-ABL PCR can be performed on the peripheral blood and represents the most sensitive test available to routinely monitor disease burden in CP-CML patients. However, substantial obstacles must be overcome (see accompanying chapter by Andreas Hochhaus, pages 128-135).6 Before PCR testing can reliably supplant BM metaphase analysis.
The ELN has defined overall response to imatinib in CP-CML as optimal, suboptimal, and failure (Table 2).4 Optimal response indicates that change in therapy is unlikely to improve survival that is projected to be close to 100% after 6-7 years. Suboptimal response means that a patient may still have substantial benefit from continuing current treatment, but with the potential for less favorable outcomes than those with optimal response. Change in therapy should be considered in this group. Failure means that favorable outcome is unlikely and that a change in therapy is indicated. These definitions are further modulated by the coexistence of warning prognostic factors that may adversely affect response to therapy and may require more stringent and careful monitoring. It is expected that these definitions will continue to evolve as longer-term follow-up provides an opportunity to evaluate EFS, PFS, and OS with greater precision.
Definitions of response in CP-CML patients treated with imatinib as defined by the ELN4

NA indicates not applicable and CHR, complete hematologic response.
Imatinib
Imatinib has been resoundingly successful at improving outcomes in CML and for much of the last decade has been the cornerstone of TKI therapy for CML. At 8 years of follow-up of the IRIS trial, the estimated OS of all patients randomized to receive imatinib was 85% and 93% when only CML-related deaths were considered.7 However, single-arm, nonrandomized trials have suggested that rates of CCyR and MMR might be improved with a higher initial dose of imatinib.8–11 To formally address this question, the Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) study randomly assigned 476 patients 2:1 to imatinib 800 or 400 mg daily. At 12 months, there was no statistically significant difference in MMR (46% vs 40%, P = .2035) or CCyR (70% vs 66%, P = .3470), but MMR rates at 3 and 6 months were higher in patients randomly assigned to imatinib 800 mg, as was the CCyR rate at 6 months (57% vs 45%, P = .0146).12 Longer follow-up is required to determine whether the faster responses achieved with higher doses of imatinib will provide clinical benefit. Nonhematologic and hematologic adverse event rates were higher in the 800-mg arm12 and, notably, whereas patients assigned to the 400-mg arm achieved an average daily dose of 388.4 mg, patients in the 800-mg arm achieved an average daily dose of only 662.0 mg, indicating the reduced tolerability of higher doses of imatinib. Nonetheless, patients who were able to tolerate higher doses of imatinib without interruption appeared to have a greater likelihood of achieving cytogenetic and molecular response. Interestingly, the German CML IV study, which used a tolerability-adapted imatinib-dose escalation scheme, found that patients who were able to tolerate 800 mg of imatinib had a significantly higher MMR rate at 12 months (59% vs 44%, P < .001).13 TIDEL I investigated higher-dose imatinib in 103 patients with newly diagnosed CP-CML using imatinib 600 mg/d with dose escalation to 800 mg/d for suboptimal response. EFS at 60 months was significantly higher in patients taking ≥ 600 mg/d compared with those who had been dose reduced to < 600 mg/d (89% vs 56%, P < .001). By 60 months, 96% of patients who had been on ≥ 600 mg/d within the first 12 months had achieved CCyR, whereas only 80% of those who had been on < 600 mg/d had achieved this milestone (P < .001). Log-rank analysis of the achievement of MMR was also significant (P = .03).14 These results suggest that patients who are able to tolerate higher doses of imatinib fare better than patients who experience unacceptable toxicity. Whether there are differences in disease biology based upon ability to tolerate higher-dose imatinib is not known.
It is important to note that criteria for dose interruptions and reductions used in clinical studies commonly consist of grade 3 or 4 nonhematologic toxicity, whereas in practical use, persistent grade 2 toxicities frequently lead to dose interruptions and discontinuations. Indeed, several studies have suggested that the outcomes with imatinib outside of the context of a clinical trial are substantially worse than those reported in the IRIS study, which may reflect in part a lower threshold of acceptable toxicity.2,15 In some instances, the ability of CP-CML patients to tolerate higher doses of imatinib may be linked to BM reserve—the capacity of the BM to rapidly reestablish karyotypically normal hematopoiesis and thereby avoid cytopenias. In addition, the German CML IV study found a benefit for 800-mg imatinib in Sokal low- and intermediate-risk CP-CML patients, but not in Sokal high-risk patients. Therefore, the higher response rates observed in patients able to tolerate higher imatinib doses may reflect both increased efficacy of more potent BCR-ABL inhibition and patient-specific factors that yield a lower likelihood of developing hematologic toxicity, thereby enabling treatment intensification in this subset of patients. Lastly, a substantial body of work suggests that imatinib is actively pumped into cells by the cationic transporter hOCT1,16–18 and several studies have provided evidence that higher doses of imatinib may be more effective at achieving clinically beneficial BCR-ABL inhibition in patients with a low level of hOCT1 expression. Despite a growing body of evidence suggesting that patients who can tolerate higher doses of imatinib can derive clinical benefit, the greater incidence of toxicity observed with higher doses of imatinib, coupled with conflicting efficacy data from randomized phase 3 studies and the cost associated with higher doses of imatinib, render imatinib doses other than 400 mg once daily investigational in newly diagnosed CP-CML.
Imatinib and IFN-α
Despite high rates of cytogenetic response with imatinib alone, major and complete molecular responses (CMRs), defined as undetectable BCR-ABL transcript by PCR, are less commonly obtained. Two studies have investigated the ability of the addition of pegylated IFN to imatinib 400 mg/d to increase the achievement of both MMR and CMR over imatinib 400 mg/d alone. The STI571 Prospective Randomized Trial (SPIRIT) randomized 636 untreated CP-CML patients to imatinib 400 mg daily, imatinib alone at 600 mg daily, imatinib 400 mg daily plus cytarabine (20 mg/m2 on days 15-28), or imatinib 400 mg daily plus pegylated IFN-α2a (90 μg weekly). The MMR rate at 18 months was 42%, 50%, 53%, and 62%, respectively (P = .004); CMR rates at 24 months were 9%, 8%, 8%, and 16%, respectively (P = .01).19 The German CML-IV study did not show an advantage of pegylated IFN plus imatinib 400 mg (34.7%) compared with imatinib 400 mg alone (3.8%) in the achievement of MMR at 12 months, but did demonstrate an increased MMR rate of 54.8% with a dose of 800 mg of imatinib daily. The apparently discrepant results with respect to the benefit of IFN added to imatinib may be related to different formulations of IFN used; in the SPIRIT study, pegylated IFN-α2a at a dose of 90 μg weekly was used, whereas the German CML-IV study added IFN-α 6 weeks after the start of imatinib at an initial dose of 1.5 million units 3 times per week and increased the dose up to 3 million units 3 times per week, depending upon tolerability. It must be noted that despite the improved molecular response rates in the imatinib plus IFN treatment arm, the SPIRIT trial failed to demonstrate improved a significant reduction in AP or BC disease transformation and death after 48 months of follow-up. The potential clinical benefit of IFN in combination with imatinib is therefore still unclear. Whereas the higher MMR rates observed with pegylated IFN-α2a are provocative, as noted below, the second-generation TKIs nilotinib and dasatinib also achieve molecular response rates that are superior to imatinib in newly diagnosed CP-CML patients and are expected to be associated with less toxicity than imatinib combined with IFN.
Second-generation kinase inhibitors in the front-line setting
The efficacy of the second-generation of BCR-ABL inhibitors dasatinib20 and nilotinib21 was initially established in imatinib-resistant or intolerant patients. However, several phase 2 studies of dasatinib22 and nilotinib23,24 have demonstrated high rates of CCyR and MMR when used in newly diagnosed CP-CML patients. In a single arm, single-institution phase 2 study of dasatinib in the front-line setting, 98% of patients achieved a CCyR and 82% achieved MMR during the course of the study.22 In a similar single-arm study at the same institution, nilotinib was associated with similar results in the front-line setting, with a cumulative CCyR rate of 98% and an MMR rate of 76%.23 A single-arm phase 2 intergroup study of nilotinib reported a 96% CCyR rate and a 85% MMR rate at 12 months.24 In a randomized intergroup phase 2 trial of first-line dasatinib 100 mg vs imatinib 400 mg, dasatinib was also associated with a superior MMR rate (59% vs 43%, P = .042) at 12 months.25
Randomized phase 3 studies have recently definitively demonstrated the superiority of both nilotinib and dasatinib in the achievement of both CCyR and MMR26,27 compared with imatinib 400 mg daily in newly diagnosed CP-CML patients. In the multinational phase 3 randomized study called Evaluating Nilotinib Efficacy and Safety in Clinical Trials-Newly Diagnosed Patients (ENESTnd), 846 patients were randomized in a 1:1:1 ratio to receive either nilotinib at a dose of 300 mg twice daily, nilotinib 400 mg twice daily, or imatinib 400 mg once daily. The CCyR rate associated with nilotinib at 12 months was 80% for the 300-mg dose and 78% for the 400-mg dose versus 65% for imatinib (P < 0.001 for both). The primary end point of MMR rate at 12 months was nearly double for nilotinib (44% for the 300-mg dose and 43% for the 400-mg dose) compared with imatinib (22%, P < 0.001 for both).27 In the multinational Dasatinib versus Imatinib Study in Treatment Naive CML Patients (DASISION) study, 519 patients with newly diagnosed CP-CML were randomized to receive dasatinib 100 mg once daily or imatinib 400 mg once daily. After a minimum follow-up of 12 months, the rate of CCyR on dasatinib was 83% versus 72% on imatinib (P = .001). The rate of MMR was also higher with dasatinib (46% vs 28%, P < .0001). Rates of discontinuation of therapy due to drug toxicity were similar between patients receiving dasatinib (5%) and imatinib (4%).26 Encouragingly, in contrast to the TOPS trial, in which the early superiority of imatinib 800 mg relative to 400 mg daily was lost by 12 months, the early superiority of both nilotinib and dasatinib relative to imatinib with respect to MMR achievement appears to be preserved with follow-up of 24 and 16 months, respectively. Whereas longer follow-up of both studies is necessary to convincingly document clinical benefit, given the relationship between depth of response and improved longer-term outcomes with imatinib therapy, the efficacy results to date have prompted the NCCN to amend its consensus guidelines to include nilotinib 300 mg twice daily and dasatinib 100 mg once daily as additional acceptable first-line treatment options for newly diagnosed CP-CML patients.5
Given the historic prognostic significance of cytogenetic and molecular responses, there is substantial optimism that the second-generation agents will convincingly improve longer-term outcomes relative to imatinib, but longer follow-up is required to definitively clarify this issue. Encouragingly, in both studies, fewer patients randomized to the second-generation drug treatment arms progressed to AP or BC disease within the first 24 months.28,29 For nilotinib, the difference is statistically significant, although there are important differences in the protocols with respect to definitions of AP transformation and time from discontinuation of study drug treatment to censorship for transformation events. In addition, due to initial concerns that nilotinib 300 mg twice daily might be subtherapeutic, the ENESTnd protocol allowed for patients with suboptimal response on nilotinib 300 mg twice daily to undergo dose escalation to 400 mg twice daily, at which time they were censored. Whether nilotinib 300 mg twice daily will retain the response durability of nilotinib 400 mg twice daily is presently an unanswered issue. Indeed, an area of considerable interest surrounds the quality of BCR-ABL kinase domain mutation that may evolve on second-generation TKIs used in the frontline setting. In the ENESTnd study, analysis of mutation evolution during the first 24 months in patients with lack or loss of MMR showed a 5-fold increase in transcript level and, at the end of treatment, encouragingly revealed a substantially lower proportion of patients having evolved mutations on the nilotinib arm compared with the imatinib arm. Of these, the absolute number of cases in which the T315I mutation was detected was equivalent. Notably, 2 of 10 patients in the nilotinib 300 mg twice daily arm who evolved new mutations demonstrated evidence of mutations not previously associated with loss of response to nilotinib, including one case of disease progression that was associated with the E459K mutation.30 A similar analysis has been performed in patients in the DASISION study, in which mutation analysis was performed in patients who discontinued for any reason (dasatinib, n = 59; imatinib, n = 64).29 BCR-ABL kinase domain mutations were detected in 10 patients in each arm; notably, 7 of the 10 mutations detected in the dasatinib arm were T315I, whereas this mutation was not detected in any of the imatinib-treated patients. Although there has been considerable historic difficulty in treating cases associated with the T315I mutation, there now appears to be an investigational TKI with considerable activity in CP-CML patients with this mutation (ponatinib; see below).
It is noteworthy that in both the ENESTnd and DASISION studies, ∼ 25% of patients in all of the treatment arms have discontinued the study drug after only 24 months. The tolerability of these 3 drugs appears comparable, and discontinuation rates due to adverse events are similar. However, many patients discontinued the study drug due to definitions of treatment failure and suboptimal response that were established by the IRIS study experience, and whether these definitions have the same prognostic significance in patients on second-generation agents used in the frontline setting remains to be determined. In addition, it is possible that the availability of more than one effective TKI for CP-CML patients in some instances led clinical investigators to remove patients from the study in an effort to pursue an alternative TKI, whereas during the IRIS study, no other highly effective medical options were available. An issue that is likely to be of greater significance when generic imatinib becomes available is that the second-generation drugs are substantially more expensive than imatinib 400 mg daily (but generally less expensive than imatinib 800 mg daily).
Bosutinib is a promising investigational second-generation TKI that, similar to dasatinib and nilotinib, is more potent in vitro and clinically vulnerable to a smaller number of resistance-conferring BCR-ABL mutations than imatinib. The multinational phase 3 BELA study compared bosutinib with imatinib in 502 previously untreated CP-CML patients, and the 12-month follow-up was reported recently.31 The primary end point, CCyR rate at 12 months (intent-to-treat) was 70% for bosutinib-treated patients and 68% for imatinib-treated patients (P = .601). MMR at 12 months favored bosutinib, 39% versus 26% (P = .002). Time to CCyR and MMR was significantly shorter with bosutinib. Trends toward lower likelihood of transformation to AP or BC CML were observed in bosutinib-treated patients, and treatment failure was significantly higher in imatinib-treated patients. However, grade 3/4 liver function test abnormalities and gastrointestinal toxicity contributed to a nearly 4-fold higher likelihood of bosutinib discontinuation, and after 12 months, only 71% of patients remained on bosutinib. As a result of the failure of this study to meet its primary end point (superior CCyR at 12 months), bosutinib remains investigational. Given the increasing prevalence of CP-CML, the number of patients who may not respond well to or tolerate any of the 3 approved TKIs is growing. Because patients can only benefit from more therapeutic options, it is hoped that bosutinib, which appears to harbor substantial efficacy, will be available to physicians who manage CP-CML patients in the near future, although its registrational path appears complex now.
Resistance and the choice of second-line therapy
The molecular mechanisms responsible for secondary resistance to imatinib have been the subject of intense study. Many of these cases involve escape of BCR-ABL inhibition, either through kinase domain mutation within BCR-ABL and resulting impairment in the ability of imatinib to efficiently bind, or presumed overproduction of BCR-ABL via genomic amplification or the acquisition of additional Ph chromosomes in the resistant clone.32–36 A subset of cases with loss of response lack evidence for one of these mechanisms and appear to be the result of BCR-ABL–independent mechanisms. By implicating poorly controlled BCR-ABL kinase activity in a substantial proportion of resistant cases, knowledge of the molecular mechanisms of imatinib resistance led directly to the hypothesis that alternative ABL inhibitors with increased potency relative to imatinib, as well as activity against imatinib-resistant BCR-ABL kinase domain mutants, would be therapeutically promising. Both dasatinib and nilotinib are more potent BCR-ABL inhibitors with activity against a broad spectrum of imatinib-resistant mutants, and both were initially FDA approved for CML patients with resistance or intolerance to imatinib. Both these approved second-generation agents are completely ineffective against the BCR-ABL/T315I mutation in vitro and clinically.
Dasatinib has activity against most imatinib-resistant BCR-ABL mutations in vitro20,37 and clinically. Notable exceptions are the highly resistant T315I mutation, which fails to respond objectively to dasatinib, and the moderately resistant F317L mutation,37 which is frequently associated with hematologic response, rarely with cytogenetic response, and occasionally with secondary resistance to dasatinib.38 Other imatinib-resistant mutations with moderate relative resistance to dasatinib include G250E and E255V/K (although many reports of achievement of CCyR have been reported in patients with these mutations) and V299L.37 In a randomized phase 3 study in imatinib-resistant or -intolerant CP-CML, of 164 patients randomized to receive dasatinib 100 mg once daily, 50% of patients achieved CCyR. The rate of PFS after 36 months within this cohort was 93%.39 Four-year follow-up of this treatment arm revealed that 45% of patients were continuing dasatinib, and 18% discontinued for disease progression, including 4% who transformed to the AP/BC phase on study. The OS at 48 months was estimated to be 82% and the EFS 66%.
Nilotinib is a structural derivative of imatinib that has also demonstrated in vitro activity against most imatinib-resistant BCR-ABL mutations with the exception of T315I, which is highly resistant.40 Nilotinib has also demonstrated clinical activity against most imatinib-resistant mutations other than T315I, and the moderately resistant mutations, Y253H, E255V/K, and F359V.21 In a phase 2 study of nilotinib with a median duration of exposure of 18.7 months (range < 1.0-36.5 months), a cumulative CCyR rate of 46% in imatinib-resistant and -intolerant patients was observed, and the cumulative MMR rate was 56%. At 24 months (n = 321), the estimated rate of OS was 87%, and EFS was 64%.41 Thirty-nine percent of patients were continuing imatinib at 24 months, and 27% discontinued for disease progression, including 3% who transformed to AP/BC on study.
The choice of which approved second-line therapy to proceed with in the case of imatinib resistance or intolerance in some cases can be informed by the presence of BCR-ABL kinase domain mutations and the individual susceptibility of these mutations to either drug. A further consideration should involve patient comorbidities, because each inhibitor has a side effect profile that may make it more or less appropriate for an individual patient (ie, increased risk of pleural effusion with dasatinib; increased risk of QT prolongation, elevations in glucose, lipase, and liver function tests with nilotinib). For patients with loss of response to nilotinib or dasatinib in the second line, a trial of dasatinib or nilotinib, respectively, is reasonable in cases that do not harbor the BCR-ABL/T315I mutation, because the other mutations that have most commonly been associated with loss of response to these agents appear not to confer cross-resistance. Whereas select mutations that are dasatinib resistant (V299L, F317I, and T315A) retain sensitivity to imatinib in vitro, mutations known to be associated with loss of response to nilotinib are highly resistant to imatinib, and imatinib therefore would not be expected to be efficacious in patients with acquired resistance to nilotinib. The ability to reliably adhere to the dosing schedules of these agents (once daily with or without food for dasatinib; twice daily, fasting, for nilotinib) may also be important considerations for select patients.
At this point, there are no studies to inform the best option for patients who develop resistance or intolerance to nilotinib or dasatinib used in the first-line setting, but a reasonable approach would be based upon the individual mutation profile and inhibitor side effects. With the exception of the BCR-ABL/T315I mutation, all mutations that confer resistance to dasatinib or nilotinib may be sensitive to the other agent, and imatinib may have activity in select cases of loss of response to dasatinib.
An area of importance concerns treatment milestones on second-generation TKIs used in the second-line setting. Given that cytogenetic and molecular responses with these agents appear to be rapidly achieved in general, evolving data suggest that failure to achieve a major cytogenetic response (≤ 35% Ph+ metaphases) within 12 months of initiation of these agents portends a poorer prognosis. It is therefore reasonable to consider allogeneic stem cell transplantation in eligible patients with lesser degrees of response on second-line TKIs who lack BCR-ABL kinase domain mutations that may explain the lack of response, although a 3-6 month trial of a different second- or third-generation TKI in an effort to achieve CCyR is reasonable. It is encouraging that available data have failed to demonstrate a negative impact of prior imatinib use on transplantation outcome, although this issue has not been adequately studied for second- and third-line agents.
Newer agents
A clear unmet need in patients who develop resistance to imatinib and the approved second-generation BCR-ABL inhibitors has been the BCR-ABL/T315I mutation, which was the first drug-resistant mutation identified,32 and is highly resistant to all 3 approved therapies. Recently, a new oral multikinase inhibitor AP24534 (ponatinib) has demonstrated significant clinical activity against T315I and other BCR-ABL mutations, as well as native BCR-ABL, in a heavily pretreated Ph+ population treated in a phase 1 clinical trial. Impressively, of 9 patients with CP-CML and the T315I mutation at study entry, 8 achieved CCyR. Of 9 CML AP/BP or Ph+ acute lymphoblastic leukemia (ALL) patients with the T315I mutation, 3 (33%) had major hematologic response, 2 (20%) had a major cytogenetic response. CCyRs were also observed in heavily refractory patients with no mutations, and patients with other mutations. Overall, 13 of 60 (22%) Ph+ patients achieved MMR, including 12 of 42 (28%) CP patients, 6 of 15 (40%) with T315I mutation confirmed at baseline, 10 of 40 (25%) patients with starting doses ≥ 30 mg. MMRs were also achieved in patients with M351T, F359C, F317L, M244V, and G250E mutations, and 1 patient with no mutation. To date, the responses in CP-CML appear to be highly durable.42 Ponatinib is currently being investigated in a phase 2 clinical trial in all phases of CML and Ph+ ALL with resistance to either dasatinib or nilotinib, and should it prove to be invulnerable to BCR-ABL kinase domain mutations clinically, it may be associated with a very low rate of loss of response and disease progression. If ponatinib is well tolerated, assessing its response rate and durability in previously untreated CP-CML patients and in advanced-phase CML and Ph+ ALL patients will be of great interest.
DCC-2036 is an allosteric inhibitor of BCR-ABL that is currently undergoing evaluation in a phase 1 study. Due to its unique mechanism of action (binding to a “switch” pocket of the kinase domain that forces the kinase to adopt an inactive conformation), DCC-2036 has promise for the treatment of a broad range of TKI-resistant BCR-ABL mutations, including T315I.43
The plant alkaloid omacetaxine is an investigational protein synthesis inhibitor that has demonstrated modest activity in a small phase 2 study of CML cases associated with the BCR-ABL/T315I mutation,44 where 10% of CP patients with the T315I mutation have achieved CCyR. Omacetaxine appears to be associated with a high degree of myelosuppression. Given its unknown mechanism of action, it is tempting to speculate that omacetaxine could represent a useful adjunctive therapy for CML patients, particularly with respect to efforts aimed at reducing or eliminating minimal residual disease, but clinical studies aimed at addressing this issue have not yet been undertaken.
Conclusions
In the past decade, imatinib has significantly improved the outlook for patients diagnosed with CP-CML. Further improvement in clinical outcomes may be expected with efforts to improve depth of cytogenetic and molecular responses and time to treatment response with newer and more potent BCR-ABL inhibitors, with the potential for the eradication of minimal residual disease in a subset of patients. The third-generation BCR-ABL inhibitor ponatinib has demonstrated significant clinical activity against the T315I mutation, the last remaining single–amino acid substitution hurdle in CML. However, important challenges remain, such as eradication of disease (cure), compound kinase domain mutations, and understanding and overriding BCR-ABL–independent mechanisms of resistance. The next decade of CML research promises to bring further improvements in clinical outcome to the point that the goal of disease control and preservation of quality of life may be achievable in nearly all CP-CML patients.
Disclosures
Conflict-of-interest disclosure: C.C.S. declares no competing financial interests. N.P.S. has consulted for Bristol-Myers Squibb, Novartis, and Ariad and has received research funding from Bristol-Myers Squibb and Ariad. Off-label drug use: the investigational agents ponatinib and DCC-2036 are discussed.
Correspondence
Neil P. Shah, Department of Hematology/Oncology, University of California-San Francisco, 505 Parnassus Avenue, Suite M1286, Box 1270, San Francisco, CA 94143-1270; Phone: (415) 514-0269; Fax: (415) 353-2467; e-mail: nshah@medicine.ucsf.edu.
