
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal hyperinflammatory condition. It may occur as a primary (genetic) condition due to mutations in genes important in the cytolytic secretory pathway that cause perforin and granzymes to induce apoptosis in target cells. Primary HLH is divided into familial HLH (FHLH1-5), in which HLH is the only manifestation of disease, and other genetic causes in which HLH is one of several clinical manifestations. The identical clinical findings may arise secondary to infectious, rheumatologic, malignant, or metabolic conditions. Whether primary or secondary, HLH therapy needs to be instituted promptly to prevent irreversible tissue damage. It is helpful to think of HLH as the severe end of the spectrum of hyperinflammatory disorders when the immune system starts to damage host tissues (immunopathology). Therefore, no single clinical feature alone is diagnostic for HLH, and it is important that the entire clinical presentation be considered in making the diagnosis. This article contains a discussion of the genetic background, clinical presentation, diagnostic dilemmas, and features that are helpful in making the diagnosis of HLH, along with a discussion of common problems in its management.
Introduction: background and pathogenesis
Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal hyperinflammatory condition caused by a highly stimulated but ineffective immune response.1 The incidence is estimated to be approximately 1.2 cases per million individuals per year,2 but this is almost certainly an underestimate. Broadly, HLH can be classified according to the underlying etiology into either primary (genetic) or secondary (acquired) HLH. Since the first description of perforin gene mutations by Stepp et al in 1999,3 significant insight has been gained into the genetic mutations that give rise to the HLH phenotype. However, to date, depending on the population group, 20%-50% of involved genes are as yet unknown4 and the pathogenesis of the condition remains to be clarified. What is clear is that all of the known genetic abnormalities lead to defects in proteins that play an important role in the cytolytic secretory pathway. In this pathway, granules containing preformed perforin and granzymes are delivered to the synaptic junction between cytolytic cells (natural killer [NK] cells and cytotoxic T cells) and their targets. Some of the same proteins are important in the transport of other granules such as melanosomes. Primary (genetic) HLH can usefully be divided into those conditions in which HLH is usually the only manifestation of the disease (FHLH1-5) and those associated with other defects such as partial albinism, although HLH may be the cause of death. Defects in PRF (FHLH2), MUNC 13.4 (FHLH3), syntaxin 11 (STX11 or FHLH4), and syntaxin-binding protein 2 (STXBP2 or FHLH5) are known to cause FHLH, whereas mutations in lysosomal transport (LYST or Chediak-Higashi), RAS-associated protein 27A (RAB27A or Griscelli 2), and adaptor protein 3 B1 subunit (AP3B1 or Hermansky-Pudlak2) can result in partial albinism and HLH (Table 1). Other inherited conditions that can present with HLH are the X-linked lymphoproliferative syndrome (XLP) and some known immunodeficiency syndromes such as X-linked SCID and X-linked hypogammaglobulinemia. Genetic testing has demonstrated that primary HLH can occur at any age, from in utero presentation with hydrops fetalis until as late as 70 years of age.
Genetic defects leading to HLH*
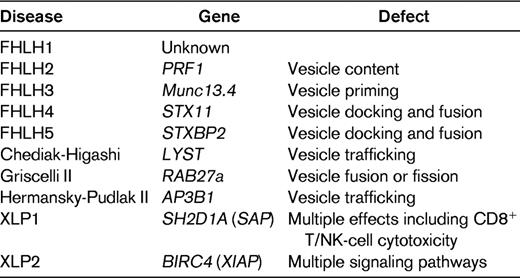
Cytotoxic cells form a conjugate with their target to form an immunologic synapse, followed by trafficking of the cytotoxic granules containing perforin and granzymes toward the immunologic synapse, docking, priming, and fusion of the cytotoxic granules with the plasma membrane. Granule content is released into the immunologic synapse and induces target-cell destruction by caspase-dependent and -independent apoptosis. Except for XLP and some congenital immunodeficiencies, all known genetic HLH is due to defects in proteins essential for this process.
HLH may also occur as a secondary disorder in association with severe infections, malignancies, rheumatologic disorders, and some metabolic diseases. Although EBV is the most common cause of infection-associated HLH, recognition that at least some of the deaths associated with avian flu and severe acute respiratory syndrome are due to HLH may lead to a reduced mortality in those conditions.5
Despite recent gains in knowledge, the pathogenesis of HLH is as yet unclear, but primary HLH is thought to involve defective termination of the immune response that results in persistent activation of macrophages and cytotoxic T cells. An alternative hypothesis involves failure to remove Ag, which results in ongoing stimulation of the immune effector cells. It is possible that both failure to clear the pathogen, resulting in continued Ag stimulation, and failure to terminate the immune response play important roles. Pathogenesis of secondary (acquired) HLH is even less clear, although patients with secondary forms of HLH are increasingly being found to have heterozygous changes or polymorphisms in the familial HLH genes. More detailed discussion of the genetic basis of HLH and the concepts underlying pathogenesis can be found in several recent reviews.4,6,7
Mortality
Before the initiation of modern therapeutic regimens, the 1-year survival rate of children with FHLH was close to 0%.8 The first organized therapeutic protocol, HLH-94, resulted in an overall survival rate of 55%, with a probability of survival of 62% for patients with FHLH after hematopoietic cell transplantation (HCT).9 With the introduction of reduced-intensity conditioning and increased experience, survival after HCT has improved to 92%.10 Reported mortalities in secondary HLH vary from 8%-22% in rheumatologic-HLH (macrophage-activation syndrome or MAS) to 18%-24% in EBV-HLH.11 Delay in diagnosis and multiorgan involvement are associated with an inferior prognosis, and whether primary or secondary, therapy needs to be instituted promptly to prevent irreversible tissue damage
Clinical features
Although the exact pathogenesis is not well understood, it is clear that the clinical manifestations of HLH are due to: (1) hyperactivation of CD8+ T lymphocytes and macrophages; (2) proliferation, ectopic migration, and infiltration of these cells into various organs; and (3) hypercytokinemia with persistently elevated levels of multiple proinflammatory cytokines, resulting in progressive organ dysfunction that may lead to death.
These interrelated factors underlie the clinical manifestations of prolonged fever, hepatosplenomegaly, bleeding, skin rash, CNS abnormalities, jaundice, and the laboratory findings of bicytopenia or pancytopenia, coagulopathy, hyperlipidemia, hypofibrinogenemia, hyperferritinemia, transaminitis, hyperbilirubinemia, hypoalbuminemia, and hyponatremia.
Diagnosis
Specific diagnostic criteria were used for patient eligibility in the Histiocyte Society (HS) trials HLH-94 and HLH-2004 (Table 2); 5 of 8 criteria are required to make the diagnosis of HLH. The utility of this has been questioned because of the lack of specificity of the various criteria.12 This reflects a lack of understanding of the point that individual criteria may lack specificity, but it is the presence of multiple criteria, reflecting the severity of the condition, that is important, along with the magnitude and progression of the abnormalities. Important examples of this are the degree of hyperferritinemia and the degree of elevation in plasma of the level of the α chain of the soluble IL2 receptor (sIL2r; sCD25) that may be seen. In a cohort of patients with elevated ferritin levels, the maximum levels seen during the hospital course was 15 830 μg/mL (range, 994-189 721) in those with HLH, but only 1356 μg/mL (range, 512-16 367) in autoimmune disease, 1120 μg/mL (range, 535-6230) in viral disease, and 972 μg/mL (range, 523-7508) in bacterial infections.13 Ferritin levels > 10 000 μg/mL were 93% specific for the HLH diagnosis. In my experience, levels > 30 000 are not uncommon in HLH and are 100% specific in the absence of an inborn error of iron metabolism. Consideration is being given to increasing the minimum ferritin level required for inclusion as a diagnostic criterion; however, patients with proven HLH may have ferritin levels only slightly above normal.
HLH-2004 diagnostic criteria
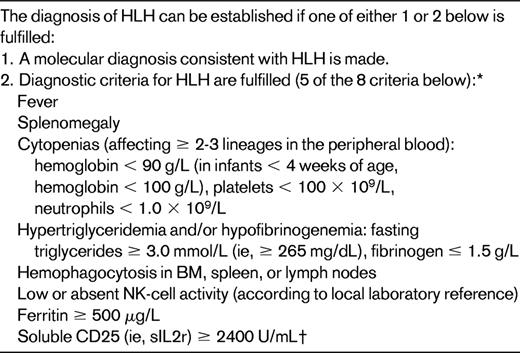
*Supportive criteria include neurologic symptoms, cerebrospinal fluid pleocytosis, conjugated hyperbilirubinemia and transaminitis, hypoalbuminemia, hyponatremia, elevated D-dimers, and lactate dehydrogenase (see text for details). The absence of hemophagocytosis in the BM does not exclude a diagnosis of HLH.
†New data show normal variation by age. Level should be compared with age-related norms.
(Adapted with permission from Henter et al20 .)
sIL2r (sCD25)
Measurement of sIL2r, reflecting the degree of activation of T cells, is useful in diagnosis and follow-up because very high levels are almost never seen outside of HLH.4,6,7 Recent work has shown age-related variations in normal levels of sIL2r,4 which are not reflected in the published criteria and need be taken into account in future studies.
Hemophagocytosis
Hemophagocytosis, a hallmark of activated macrophages, is neither specific nor sensitive for HLH and its presence should only be considered supportive
NK function
Demonstration of very low NK function is helpful in the HLH diagnosis. However, like sCD25 and sCD163 levels,14 measurement of NK function requires the sending of samples to specialized laboratories and these results are not always available to help with a timely diagnosis. In addition, even patients with severe FHLH may have normal NK function, so normal NK function should not preclude the diagnosis of HLH. Patients may therefore have to be diagnosed with HLH in the absence of 5 criteria and patients with 5 criteria (with the levels as published) do not always have HLH. Therefore, the entire clinical presentation needs to be taken into consideration when making a diagnosis of HLH and good clinical judgment is essential. Thinking of HLH not as a specific rare diagnosis, but as the severe end of the spectrum of the hyperinflammatory syndromes, when the hyperimmune response causes damage to the body, is helpful in this regard. To reflect this, it has been suggested that the criteria be revised to include a category designated “Immunopathology” (evidence of pathologic inflammation) (Jordan M, personal communication). In this model, the finding of evidence of immunopathology is essential for the HLH diagnosis. Evidence of immunopathology includes liver dysfunction, and in particular evidence of portal triaditis, the consumptive coagulopathy that underlies in part the cytopenia and bleeding problems, the CNS dysfunction shown to be present in 62% of patients in the HLH-94 study,15 and the progression to respiratory and kidney failure that may occur. Helpful findings that are not part of the published criteria are the presence of conjugated hyperbilirubinemia (due to the tropism of histiocytes to the biliary tree), transaminitis, hypoalbuminemia, hyponatremia, high D-dimers even when the international normalized ratio and partial thromboplastin time are normal, and CNS pleocytosis. Other evidence of ongoing consumption, such as a poor response to transfusions, may help to distinguish HLH-induced cytopenia from cytopenia due to other causes in the absence of HLH.
Because the liver is the most commonly affected organ, diagnosing HLH in the absence of at least increased transaminases is problematic. It has also been suggested that the diagnosis should be considered in all patients presenting with idiopathic liver failure. A recent retrospective study at The Hospital for Sick Children in Toronto found evidence of HLH in 33% of children presenting with liver failure of unknown etiology (S.W., unpublished data; Figure 1A-B). The retrospective nature of the study is problematic and a prospective multi-institutional study is important. If true, however, timely therapy may prevent the need for liver transplantation in some patients.

Evidence of HLH in children presenting with liver failure of unknown etiology. (A) BM aspirate showing hemophagocytosis in a patient admitted with acute liver failure. (B) Pathology from the explanted liver showing extensive necrosis and infiltration with histiocytes (arrow).
Evidence of HLH in children presenting with liver failure of unknown etiology. (A) BM aspirate showing hemophagocytosis in a patient admitted with acute liver failure. (B) Pathology from the explanted liver showing extensive necrosis and infiltration with histiocytes (arrow).
New laboratory techniques
sCD163
CD163, a receptor for hemoglobin-haptoglobin complexes, is a marker for activation of alternative-pathway scavenger macrophages. The plasma levels of soluble CD163 in HLH are considerably higher than those found in infections, autoimmune diseases, and cancer.14 Correlation with fatal outcome has been shown in patients with liver disease and sepsis.14,16,17 The combination of sCD25 (produced by activated T cells and dendritic cells) and sCD163 (activated macrophages) may be useful in the diagnosis and follow-up of HLH disease activity.16
In addition to the measures of hypercytokinemia such as the sCD25 and SCD163 levels described above, new and rapid laboratory techniques have become available for screening for some of the genetic mutations associated with HLH. To protect the cytotoxic cell from preformed perforin and granzymes, the granules are lined with LAMP-1 (CD107a), which can be measured on the cell surface after degranulation by flow cytometry. The absence or decreased intensity of CD107a signaling therefore screens for a genetic cause of failure of degranulation.18,19 Perforin protein expression is also measurable by flow cytometry, as is the protein product of the 2 XLP genes. The finding of deficiency of these proteins helps to support the diagnosis of HLH in these conditions.11 Because the finding of disease-associated genetic mutations can help to confirm the diagnosis in difficult cases of HLH, it is recommended that CD107a screening and perforin protein expression be done in all such patients. All male patients with EBV-HLH should be screened for the XLP proteins, and in those patients with persistent or recurrent disease, sequencing of the FHLH and XLP genes should be done. The need for HCT in all patients with primary HLH and the importance of genetic counseling and screening of siblings suggests that most, if not all, young pediatric HLH patients should undergo full genetic testing. However, the financial implications need to be considered before that recommendation is extended to all patients with HLH.
In summary, recognition that it is the magnitude and progression of the entire spectrum of abnormalities, together with the utilization of the wide range of laboratory tests which indicate the hyperinflammatory state, that will help with this difficult diagnosis. It is important to remember that the diagnosis needs to be made early and therapy instituted as promptly as possible in severe HLH to prevent irreversible damage and death.
Treatment of HLH
Effective early therapy reduced the mortality from HLH from 95% to ∼30%-35% in the HLH-94 trial.7 Initial therapy consists of combinations of immunosuppressive therapy and pro-apoptotic chemotherapy.4 Despite the early use of chemotherapy, only when etoposide, a proapoptotic chemotherapy drug, was added were sustained remissions seen. The HLH-94 and HLH-2004 trials used high-dose dexamethasone, etoposide, and cyclosporine A (CSA), together with intrathecal methotrexate (IT MTX) for patients in whom CNS-HLH did not remit after 2 weeks of dexamethasone.20 A concurrent French trial using prednisolone, antithymocyte globulin, and CSA appeared to induce responses in a higher number of patients, and with possibly less toxicity, but had a higher relapse rate,21 and all patients required IT MTX after correction of the coagulopathy. A pilot trial using a combination of etoposide, dexamethasone, antithymocyte globulin, and IT MTX has just opened (Jordan M, personal communication).
All primary HLH patients need HCT for cure, with survival after myeloablative-conditioning HCT ranging from 50% for haploidentical donor HCT to 70% for family donor HCT.7,22,23 In patients with apparent primary HLH but no known genetic mutation, decisions about the suitability of family members to act as donors may be difficult. It is well known that family members with identical mutations may present at very different ages, likely due to the need for an environmental stimulus (such as EBV) to precipitate the clinical HLH. In probands with very low NK function, assessment of the NK function in a potential sibling donor may be helpful.
Reduced-intensity conditioning with fludarabine, melphalan, and alemtuzumab (CAMPATH) has been recently shown to reduce the high transplantation-related mortality associated with myeloablative conditioning (35% at day 100) and to significantly improve survival,10 but problems with primary graft failure and loss of chimerism resulting in a requirement for donor lymphocyte infusions have limited the type of donor suitable for reduced-intensity conditioning HCT. What is apparent in HLH HCT is the significant learning curve associated with these procedures, which should be done at centers with experience in HLH HCT.
Patients going into transplantation in good remission had a better chance for survival in most, but not all, studies. Patients with FHLH not achieving remission should undergo HCT as soon as a donor is available because they still have an ∼ 50% chance of survival after HCT.16 It is controversial whether a sibling who is found to have the same genetic mutation but is as yet asymptomatic should undergo HCT. It is believed that all siblings with homozygous mutations will eventually develop clinical HLH, although the time interval may vary. Going into HCT before any organ damage may reduce posttransplantation complications; however, a group of physicians at a recent HS meeting unanimously agreed that they would not submit a sibling to HCT unless they had had clinical HLH. This may be a matter for individual transplantation centers to decide.
Other special issues related to HLH management
Except for rheumatologic-HLH (MAS), all forms of HLH can be initially treated on the same protocol, and therefore there is no need to distinguish primary from secondary HLH at the time of diagnosis. There should be no delay in starting therapy for this purpose. Eventually, the distinction does need to be made because of the requirement for HCT for all primary HLH patients. If none of the known genetic causes of HLH are found, an as-yet-unknown genetic defect may still be present. In this case, the decision to send a patient to HCT is based on whether the disease reactivates after adequate therapy. In that case, a genetic basis for disease is presumed.
In patients with primary HLH who have very low NK function, the carriers may have intermediate low levels of NK function.24 This may be useful in diagnosis and also in monitoring a younger sibling after birth. If the infant develops normal NK function (usually by 3 months of life), he/she is unlikely to have the disease.
Not all patients with HLH need to be started on the full protocol. Patients with MAS (rheumatology-associated HLH) may respond to corticosteroids alone or a combination of corticosteroid and CSA and /or IV gammaglobulin. Recent therapies for MAS including TNF-inhibiting agents, IL1 inhibitors/antibodies, and anti-IL6 antibodies have been useful in some MAS patients, but MAS may also be precipitated by these agents.4
Corticosteroid therapy alone may be tried for other potentially secondary cases of HLH, and some patients do respond to monotherapy; however, the treating physician must be prepared to move very quickly (within 24-48 hours) to full therapy if the disease does not respond rapidly or progresses.
As briefly discussed above, etoposide is a very useful anti-HLH drug and appears to also have specific activity in EBV-HLH. In my experience, some patients with HLH and even a minority with MAS do not respond until etoposide is added. Previous concern regarding the use of etoposide in patients with high levels of conjugated bilirubin appear to have been overstated, and full-dose etoposide can be used unless renal function is also abnormal.
EBV-HLH
EBV infection is the most common cause of infection-associated HLH, and may trigger HLH in patients with familial disease. EBV-HLH may respond to steroids alone, or patients may require more aggressive therapy including etoposide or even HCT for cure. In a large Japanese series, survival was significantly better if etoposide-containing therapy was initiated early and if > 4 doses of etoposide were given.25 It has been suggested that etoposide use is “inevitable” in very severe or refractory EBV-HLH, but also that the cumulative dose should be kept < 3000 mg/m2 to minimize the risk of secondary AML.11 Another useful addition to therapy is rituximab, which can eliminate B cells in which EBV proliferates.26 In EBV-HLH, however, EBV has been shown to also proliferate in T and NK cells9,27 and serial quantitative PCR of EBV DNA may show recurrence of the EBV load despite the absence of CD20+ B cells. In this situation, alemtuzumab may be beneficial.4,11 Some patients with HLH after primary EBV infection later develop aggressive recurrent EBV-HLH requiring retreatment and HCT, despite not demonstrating abnormalities in the known XLP genes, signaling lymphocyte activation molecule associated protein (SAP or SH2D1A) and X-linked inhibitor of apoptosis (XIAP or Birc4).
MA-HLH
Lymphoma is the most common cause of malignancy-associated HLH (MA-HLH). It is more common in adults than in children, which may reflect the fact that HLH has been reported primarily with lymphoma or leukemia of peripheral T- or NK-cell lineages. In Japan, EBV-positive NK/T lymphoma-associated HLH (LA-HLH) occurs mainly in young adults, whereas EBV-negative B-cell LA-HLH is seen in patients older than 40 years.28 Whereas MA-HLH has been reported with many different pediatric malignancies, in my experience, the most common associations are with anaplastic and nonanaplastic peripheral T-cell lymphomas often involving the skin and with leukemia, including acute monoblastic leukemia. We and others have described HLH occurring during induction therapy for B-precursor acute lymphoblastic leukemia (ALL),29,30 and a review of the literature found 24 cases of HLH during and after therapy for B- and T-precursor ALL.30 Several case reports of ALL presenting with HLH as the first manifestation of leukemia have been published, including a recent report of 2 patients with a heterozygous MUNC 13.4 mutation and a heterozygous perforin (PRF) gene mutation, respectively, who developed ALL within 1-4 months after therapy for HLH,31 leading to speculation that abnormalities in these genes may increase the risk of malignancy.
Delay in diagnosis of the malignancy is common in MA-HLH and the reported prognosis in adults is poor. Patients usually require HLH-specific and malignancy-specific therapy. It has been suggested that high-dose chemotherapy and HCT may improve survival in LA-HLH.9
Salvage therapy
Good supportive care, including the removal of cytokines with plasma exchange or even exchange transfusion in the very young infant, has helped to maintain patients until other therapies have a chance to work. Other salvage therapies include high-dose pulse corticosteroids and/or the anti-CD52 antibody alemtuzumab. Alemtuzumab suppresses T cells and histiocytes and has proven to be a successful salvage strategy,4,32 but careful surveillance for opportunistic infections such as CMV is essential. Other published salvage therapies include antithrombin III,32 infliximab (anti-TNF antibody),33 and daclizumab (anti-CD25 antibody).34 In MAS patients, IL1 and IL6 inhibition have been successful in some but not all patients. The role of these therapies in other HLH patients has not been established. Failure of salvage regimens should result in the strong consideration of HCT.
Recurrence of signs of inflammation on therapy
Persistent or recurrent fever may represent ongoing HLH or opportunistic infections. A secondary infectious process must be eliminated before immunosuppressive therapy is intensified. Fungal infections in particular represent an important cause of death in this population.35 If an opportunistic infection is not found, it is often helpful to first increase the doses of drugs such as steroids and etoposide. If needed, other salvage strategies as outlined above should be tried.
Adult HLH
Although FHLH is reported in adults, most adult HLH is likely to be secondary to an underlying disease. Japanese data suggest that the most common cause of secondary HLH in adults is LA-HLH. There are few data directly comparing prognosis in adult and pediatric cases, but the prognosis is poor in LA-HLH in adults,28 and a study of EBV-HLH in Japan showed that the outcome was significantly worse in adult patients.36 Whether this reflects late diagnosis and therapy is unknown. In general, the same therapy is recommended for adult HLH as for the pediatric patients, but rapid move to more aggressive protocols (within days, not weeks) and even HCT should be emphasized for nonresponding patients.
Conclusion
HLH is a potentially fatal condition is often missed in children and adults. Goals for the future include increasing awareness of the condition, which requires both early diagnosis and early effective therapy to further reduce mortality.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: all drugs used in HLH are off-label, including newer therapies such as alemtuzumab, infliximab, daclizumab, and IL1 and IL6 antagonists.
Correspondence
Sheila Weitzman, MB, Division of Hematology/Oncology, The Hospital for Sick Children, 555 University Avenue, Toronto, ON, Canada M5G1X8; Phone: (416) 813-5872; Fax: (416) 813-5327; e-mail: sheila.weitzman@sickkids.ca.
