
Abstract
Myelofibrosis (MF), either primary or arising from previous polycythemia vera (PV) or essential thrombocythemia (ET), is the worst among the chronic myeloproliferative neoplasms in terms of survival and quality of life. Patients with MF have to face several clinical issues that, because of the poor effectiveness of medical therapy, surgery or radiotherapy, represent largely unmet clinical needs. Powerful risk stratification systems, applicable either at diagnosis using the International Prognostic Scoring System (IPSS) or during the variable course of illness using the Dynamic International Prognostic Scoring System (DIPSS) and DIPSS Plus, allow recognition of categories of patients with survival times ranging from decades to < 2 years. These scores are especially important for therapeutic decisions that include allogeneic stem cell transplantation (allogeneic SCT), the only curative approach that still carries a nonnegligible risk of morbidity and mortality even with newest reduced intensity conditioning (RIC) regimens. Discovery of JAK2V617F mutation prompted the development of clinical trials using JAK2 inhibitors; these agents overall have resulted in meaningful symptomatic improvement and reduction of splenomegaly that were otherwise not achievable with conventional therapy. Intriguing differences in the efficacy and tolerability of JAK2 inhibitors are being recognized, which could lead to a nonoverlapping spectrum of activity/safety. Other agents that do not directly target JAK2 and have shown symptomatic efficacy in MF are represented by inhibitors of the mammalian target of rapamycin (mTOR) and histone deacetylases (HDACs). Pomalidomide appears to be particularly active against MF-associated anemia. However, because these agents are all poorly effective in reducing the burden of mutated cells, further advancements are needed to move from enhancing our ability to palliate the disease to arriving at an actual cure for MF.
Introduction
The term myelofibrosis (MF) refers to primary MF (PMF)1 and to the phenotypically overlapping conditions that develop as (usually) late evolution of either polycythemia vera (PPV-MF) or essential thrombocythemia (PET-MF).2 Most information available in the literature concerns PMF, and rigorously defined criteria for risk stratification have been developed specifically for PMF. Conversely, because manifestations and therapeutic approaches are virtually the same in PMF and PPV/PET-MF, no distinction among them will be made in this review. MF is the most symptomatic and has the worst prognosis among the Philadelphia-chromosome–negative chronic myeloproliferative neoplasms (MPNs). Among the 1054 PMF patients reported by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT), the median survival was 69 months (95% confidence interval [95% CI] =61-67 months). Causes of death were leukemia in 31%, “accelerated” phase without leukemia transformation in 19%, thrombosis in 14%, bleeding in 5%, infections in 10%, portal hypertension in 4%, second neoplasia in 4%, and other causes in 10%.
Diagnosis of MF
Current diagnosis of PMF is based on the 2008 World Health Organization (WHO) criteria,1 which enlist histopathologic, morphologic, clinical, and molecular-cytogenetic variables. Histopathology is key to the diagnosis, demonstrating the atypia of megakaryocyte proliferation even in the absence of overt reticulin fibrosis; it also helps in the differential diagnosis with nonclassical MPN disorders, myelodysplastic syndromes including the del(5q) syndrome, PV or ET with initial BM fibrosis, and other myeloid neoplasms. A second major diagnostic criterion is represented by the detection of JAK2V617F or a MPL mutation, occurring in 60% and 5%-10% of the patients, respectively, and other MPN-associated molecular abnormalities (eg, CBL, ASXL1, TET2, and EZH2) or clonal markers (particularly trisomy 9 or 13q−) that distinguish PMF from reactive marrow fibrosis.3 The absence of BCR/ABL rearrangement allows the exclusion of chronic myelogenous leukemia. Minor criteria are represented by leukoerythroblastosis, raised serum lactate dehydrogenase levels, anemia, and splenomegaly.
One major point of debate concerns the identification of “prefibrotic” (or early) MF with thrombocytosis in the settings of differential diagnosis with typical (or “true”) ET. The validity of such a distinction based on purely histopathologic criteria has been questioned,4,5 but 2 recent studies highlighted clinically meaningful differences between ET, strictly defined according to the WHO criteria, and prefibrotic MF.6,7 Compared with ET, patients with prefibrotic MF had reduced overall survival, higher rates of leukemic transformation, and overt fibrotic progression.6
Post-PV and post-ET MF is diagnosed according to IWG-MRT criteria.2 The diagnosis requires as a major criterion the development of BM fibrosis of grade 2-3 (or 3-4, depending on the scale) and at least 2 minor criteria: leukoerythroblastosis results, increasing splenomegaly, and the development of ≥ 1 constitutional symptom(s) for PV and ET; new-onset, therapy unrelated anemia or reduced phlebotomies for PV; anemia and a decreased hemoglobin level of ≥ 2 g/dL from baseline, and increased serum lactate dehydrogenase levels for ET. The term “blast-phase PMF” is currently used for leukemic transformation.8
IPSS/DIPSS/DIPSS plus allows reliable patient risk stratification
MF remains an incurable disease for patients who are not successful recipients of allogeneic stem cell transplantation (allogeneic SCT), because no other medical intervention has been shown to improve survival to date. Therefore, treatment is largely risk and problem oriented according to each individual patient. A powerful risk-stratification system, the IPSS, was developed by the IWG-MRT using a multi-institutional series of 1001 patients with PMF.9 This system employs 5 variables, estimated at diagnosis, for prediction of survival: (1) age > 65 years, (2) hemoglobin level < 100 g/L, (3) leukocyte count > 25 × 109/L, (4) ≥ 1% blasts in the peripheral blood, and (5) the presence of constitutional symptoms (eg, night sweats, loss of > 10% body weight in the last 6 months, noninfectious fever) (Table 1). Projected survival ranged from 135 months (95% CI = 117-181 months) in the low-risk category (no risk factors) to 95 months (95% CI = 79-114 months) in the intermediate-1 category (one risk factor), 48 months (95% CI = 43-59 months) in the intermediate-2 category (2 risk factors), and 27 months (95% CI = 23-31 months) in the high-risk category (3 or more risk factors). Considering that modification of the disease over time, with acquirement of additional risk factors not present at diagnosis, could profoundly affect survival, the IWG-MRT subsequently developed the DIPSS,10 relying on the data obtained in a cohort of 525 PMF patients with extensive follow-up information. The DIPSS includes the same 5 variable as the IPSS, but the acquisition of anemia was assigned a score of 2 because this event affected survival with an hazard ratio roughly double the other parameters. In this series, 63% of the patients became anemic at 15 years from diagnosis, 22% acquired the risk variable “leukocytosis,” 39% acquired “blasts ≥ 1%,” and 22% developed constitutional symptoms. Therefore, the advantage of the DIPSS score is that it permits a tailored framework for clinical decision-making at any time during the disease course.
Prognostic score systems for patients with PMF
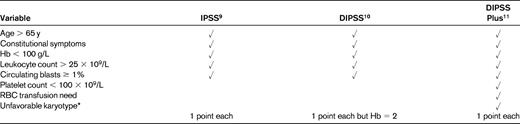
Hb indicates hemoglobin.
*Unfavorable karyotype indicates any of the following: +8, −7/7q−, i(17q), inv(3), −5/5q, 12p−, or 11q23 rearrangements.
Additional factors not included in the IPSS that affect survival are represented by red cell transfusion need,12 thrombocytopenia,13 and “unfavorable” karyotype.14 Regarding the latter, patients with unfavorable karyotype, which includes a complex karyotype or sole or 2 abnormalities such as +8, −7/7q−, i(17q), inv(3), −5/5q−, 12p−, or 11q23, had a median survival of 2 years compared with 5.2 years for those with a “favorable” karyotype, defined as no abnormality or any other apart from those included in the above category, the 5-year survival rates were 8% and 51%, respectively. The newly devised DIPSS Plus score11 incorporates these additional 3 variables for improved prognostic categorization (Table 1). In a series of 793 patients, median survival times were 185, 78, 35, and 16 months for the low, intermediate-1, intermediate-2, and high-risk categories, respectively.
A negative prognostic role for survival in PMF has been attributed also to a low (≤ 25%) V617F allele burden,15,16 nullizygosity for the JAK2 46/1 or “GGCC” haplotype,17 increased circulating plasma levels of cytokines (IL-8, IL-2R, IL-12, and IL-15),18 and a monosomal karyotype.19 However, these prognostic variables remain to be validated prospectively and should not be considered routine tests at this time.
Treatment is guided by risk stratification and the patient's clinical needs
The lack of disease-curing “conventional” therapies (apart from allogeneic SCT) and the multifaceted and ever-changing manifestations largely dictate a “clinically oriented” therapeutic approach to MF. According to the recommendations from the European Leukemia Net (ELN), “the main goals of therapy in PMF are prolongation of survival and, if possible, also cure, which is currently only achieved by SCT. If prolongation of survival or cure is not possible, symptom-orientated palliation and quality of life are the main goals.”20
The most common clinical issues that MF patients have to face are listed in Table 2. These include anemia (either moderate or transfusion dependent), splenomegaly and/or hepatomegaly, the development of foci of nonhepatosplenic hematopoiesis, myeloproliferation manifesting with marked leukocytosis or thrombocytosis, an increased risk of thrombohemorrhagic complications, and a spectrum of debilitating constitutional symptoms. Therefore, before embarking upon a treatment decision, one should accurately determine which are the immediate needs of the patient, the overall performance status, the potential benefits and risks of conventional treatment(s), the availability of novel therapies, and last, but not least, the patient's wishes. As a matter of fact, risk-stratification criteria such as the IPSS and its modifications do not entirely account for the heterogeneity of hematologic and clinical manifestations that may require a treatment. For example, a low-risk subject can suffer from extensive thrombocytosis or massive splenomegaly that call for therapeutic intervention. Therefore, IPSS risk stratification is fundamental in determining whether the patient belongs to the high-risk or intermediate-2 category to evaluate the appropriateness of considering SCT. However, if this is not the case or if the procedure is refused by the patient, then treatment is largely risk-stratification independent and should aim instead at addressing the unique patient's clinical needs.

Allogeneic SCT: cure for whom, when, and at what cost?
The achievement of a JAK2V617F negativity after SCT is associated with complete hematologic and cytogenetic remission and results in a low incidence of relapse,21 providing proof-of-concept that cure is achievable through elimination of the mutated clone. Reduced-intensity conditioning (RIC) regimens have been used in recent years with the dual aims of improving the high transplantation-related mortality rate of conventional-intensity conditioning (CIC) regimens and to accommodate the advanced median age of MF patients. However, no prospective study comparing RIC with CIC, or SCT with conventional therapy, has yet been completed. Furthermore, published series have included patients with a wide age range and heterogeneous clinical characteristics, prognostic risk categories, conditioning regimens, sources of donor stem cells, and types of immunosuppression after transplantation. Analysis of the largest published series indicated that transplantation-related mortality ranged from 10% to more than 40%, overall survival at 3 years was between 30% and 50%, and grade II-IV acute GVHD occurred in 20%-60%. The incidence of extensive chronic GVHD is not well established, but in some series it affected more than two-thirds of the patients. Variables associated with transplantation outcome are represented by HLA disparity, use of an unrelated donor, age of the recipient, and a transfusion history of > 20 red blood cell units.22 The significance of a massively enlarged spleen regarding transplantation outcome is debated,22,23 and splenectomy cannot be routinely recommended in preparation for SCT. However, is it reasonable to explore the safety and efficacy of novel drugs, such as JAK2 and mTOR inhibitors, that produce rapid spleen shrinkage and improvement of constitutional symptoms in the immediate pretransplantation setting.
In summary, no evidence-based recommendation can be provided at this time about who should certainly undergo SCT and when. Indeed, a retrospective analysis of young (< 60 years), high-/intermediate-risk PMF patients who were managed with nontransplantation procedures at 3 experienced centers reported 3-year survival estimates of 55%-77%, overall comparable to the results obtained with either CIC or RIC SCT.24 Therefore, although the ideal candidate for SCT is the patient with IPSS intermediate-2 and high-risk disease at diagnosis with a preference for CIC if younger than 40-50 and RIC if older, a decision to refer this patient to SCT is commonly postponed in clinical practice. Conversely, patients who acquire additional unfavorable cytogenetic aberrations (particularly of chromosome 17) and/or manifest signs of disease progression (eg, increasing blasts and worsening of thrombocytopenia) that could imply impending transformation to acute leukemia and that predict survival at < 1 year25 should be subjected to SCT without further delay. An open, wise, and detailed discussion with the patient of the different issues associated with SCT, including the hematologist working in the transplantation unit, is warranted and the decision should be shared as much as possible.
Management of clinical needs for non-SCT candidates with conventional therapies
Patients who have no or minimal symptoms, irrespective of age and risk category, should be managed with a “watch-and-wait” strategy, postponing a therapeutic decision until the appearance of hematologic abnormalities or clinical manifestations that require treatment and/or cause a shift in risk category according to the DIPSS/DIPSS Plus score. In high-risk patients, strict follow-up is especially intended to define the optimal timing for SCT, if feasible.
Anemia
The list of conventional agents used to treat PMF-associated anemia is remarkably long (Table 3), which is an indirect measure of their substantially low or at best unpredictable efficacy. Low-dose prednisone, erythropoiesis-stimulating agents (ESAs), androgens, danazol, and thalidomide are commonly used, and often in that order.26,27 ESAs are worth trying in patients with moderate, non-transfusion-dependent anemia and a low (< 125 U/L) serum erythropoietin level; a rapid enlargement of the spleen under treatment has occasionally been reported. Response rates vary from 20%-60% in different studies, with no clear support for darbepoetin-alpha versus conventional recombinant erythropoietin28 ; indeed, no prospective randomized study of the value of ESAs has been published. Responses are usually short-lived (< 1 year). ESAs are not indicated in anemic subjects with established transfusion dependency. If ESAs do not work or lose efficacy and there are no contraindications, different androgen preparations or danazol can be used; responses may occur in ∼ 20%, often after > 3 months of treatment. Low-dose (50 mg/d) thalidomide in association with tapering prednisone has produced responses in anemia in ∼ 20%-40% of patients29 ; however, this treatment is often poorly tolerated, with peripheral neuropathies, constipation, and somnolence leading to discontinuation of the drug in most. Due to the risk of thrombosis, prophylaxis with aspirin is recommended in patients with a platelet counts > 50 × 109/L.
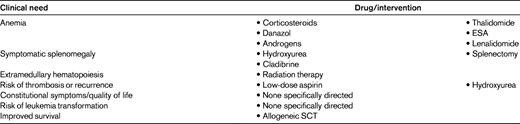
In the infrequent PMF patient with del(5q31)–associated anemia, lenalidomide is the recommended first-line therapy because significant improvement, with resolution of anemia and occasionally evidence of molecular remission, has been reported.30 Conversely, lenalidomide in association with prednisone appeared to be of little benefit in improving anemia (19%) or reducing splenomegaly (10%) in a multicenter Eastern Cooperative Oncology Group phase 2 trial in PMF patients lacking the del(5q); furthermore, treatment was poorly tolerated with grade 3/4 myelosuppression and nonhematologic toxicity in 88% and 45%, respectively.31 Slightly better results, with 30% and 42% responses in anemia and splenomegaly, respectively, but a comparable toxicity profile, were reported in another trial. A reduction of the JAK2V617F allele burden and BM fibrosis has been documented in some patients.32 At present, lenalidomide in association with prednisone exclusively is recommended for patients with the del(5q31) abnormality.
Splenomegaly
Development of massive splenomegaly with a variety of associated problems is the most common symptom and the most formidable therapeutic challenge.33 Splenomegaly-related manifestations include pain, early satiety, bloating, splenic infarctions, signs due to compression of abdominal organs or portal hypertension, and cytopenias. The main approaches for the treatment of symptomatic splenomegaly are medical (with oral or IV drugs) or surgical. The first choice is hydroxyurea, which usually produces modest responses at higher doses, which are not easily tolerated because of newly developed or exacerbated cytopenias. Greater than 25% and 50% reductions in spleen size have been reported in up to 35% and 17%, respectively, of the patients treated with hydroxyurea.33 These responses are usually modest and transient, occur mostly in subjects with nonmassive (< 10 cm) splenomegaly, and preferentially in those who are JAK2V617F mutated.26 In the randomized COMFORT-II study (described below), none of the 73 patients in the “best-available treatment” arm, of whom 60% received hydroxyurea, achieved a > 35% spleen volume reduction (which equals a 50% reduction in palpable spleen).34 Busulfan or melphalan are used in older subjects who do not tolerate or do not respond to hydroxyurea, but they are even more myelosuppressive. Responses in splenomegaly with low-dose (50 mg/d) thalidomide are infrequent (< 20%). In cases of massive, refractory splenomegaly monthly IV cladribine (2-CdA; 2-chlorodeoxyadenosine) courses produced up to 50% responses, with severe but reversible cytopenias being the main toxicity.35
Splenectomy remains a valuable alternative to medical therapy when the latter is ineffective or not tolerated. Indications for splenectomy are symptomatic massive splenomegaly, symptomatic portal hypertension with esophageal varices and/or bleeding, profound cachexia, and transfusion-dependent anemia Isolated thrombocytopenia is not an indication for splenectomy. Removal of the spleen produces improvements in mechanical symptoms, especially early satiety and pain, in most cases and is often followed by weight gain in cachectic patients, but it is usually not as effective against other constitutional symptoms. Improvement in anemia and thrombocytopenia has been reported in 50% and < 30% of patients after splenectomy, respectively. Perisurgical complications occur in up to 30%-50% and are fatal in 5%-10% of patients. Careful assessment of potential candidates, exclusion of comorbidities or ongoing coagulopathy, and a detailed discussion with the patient of expected benefits and potential risks are all mandatory. Referral to a surgeon experienced in removing these unusually large spleens and used to working in collaboration with the referring hematologist is a winning strategy. Complications of splenectomy include bleeding, thrombosis, infections, and a “myeloproliferative” reaction, with thrombocytosis and leukocytosis that can usually be well controlled with hydroxyurea and do not necessarily imply disease progression. It is prudent to lower the platelet count to ∼ 200 × 109/L in the preoperative setting and to maintain it around this level in the postoperative period with hydroxyurea or platelet apheresis (in cases of unusually extreme thrombocytosis) to reduce the risk of thrombosis. Prophylactic full-dose heparin should be delivered for at least 4-6 weeks. Not infrequently, thrombosis occurs in the splanchnic veins; therefore, ultrasound of the abdomen vessels should be performed 7 and 30 days after the procedure to detect this complication early and to maximize anticoagulation if necessary. Progressive hepatomegaly sometimes follows splenectomy, likely due to the migration of hematopoiesis, and a markedly enlarged liver is a contraindication to splenectomy. Current data do not convincingly support an increased rate of leukemic transformation after spleen removal.
Although radiotherapy of an enlarged spleen may potentially provide some relief of symptoms due to mechanical discomfort, this approach should only be used in very selected cases that are unresponsive to conventional treatment and cannot undergo (or refuse) splenectomy. The recommendation is made for the following reasons: (1) responses last a few months at maximum and usually do not even produce satisfactory relief of symptoms, (2) radiation can induce marked and durable cytopenias responsible for 10%-15% mortality, and (3) radiation causes local fibrosis with splenic adhesions to surrounding tissues that make a subsequent splenectomy technically more complicated and increase the morbidity and mortality of the procedure.33 Hepatic and abdominal radiation may palliate hepatomegaly and/or ascites, but cytopenias are common and responses are brief.
Extra-hepatosplenic hematopoiesis
Hematopoiesis in sites and organs other than the spleen and liver most often involves the peritoneum with ascites, the lungs with pulmonary hypertension and pleural effusions, the lymph nodes with lymph adenomegaly, the thoracolumbar vertebral column with pain or neurologic defects due to compression, and the lower and upper extremities with analgesic-refractory pain. Asymptomatic localizations are left untreated; low-dose, fractionated (up to 1 Gy in 10 fractions) radiation in involved fields is the treatment of choice for symptomatic manifestations. Dyspnea, cough, shortness of breath, and lower-extremity edema raise the suspicion of diffuse lung parenchymal involvement, which can be substantiated by measuring the systolic pulmonary artery pressure and/or a positive technetium 99m sulfur colloid scintigraphy. Single-fraction (100 cGy) whole-lung radiotherapy induces prompt regression of symptoms. Surgical removal of extra-hepatosplenic foci of hematopoiesis is required only occasionally.
Risk of thrombosis
Patients with PMF suffer from an increased risk of major cardiovascular events. Fatal and nonfatal thromboses were reported in 7.2% of 707 PMF patients included in a multi-institutional series, with a rate of 1.75% patient-years not dissimilar from that reported in ET (1%-3% patient-year).36 Risk factors for thrombosis were age > 60 years and a JAK2V617F mutational status, particularly if the latter was associated with leukocytosis. Whether the risk of thrombosis could be favorably affected by hydroxyurea, as in PV or ET, is unknown; however, even if cytotoxic treatment is not otherwise required for manifestations of the underlying disease, hydroxyurea and low-dose aspirin should be prescribed in older patients and in those with a history of thrombosis.
Constitutional symptoms
Efficacy of conventional therapies against severe constitutional symptoms is modest at best, although in one study up to 80% of the patients receiving hydroxyurea had a response in constitutional symptoms.37 Indeed, results from the randomized COMFORT-II study indicated that none of the patients in the best-available therapy arm presented measurable improvements in symptoms, as measured by the EORTC QLQ-C30 or FACT-Lym scores.34 Low-dose prednisone may sometimes produce a feeling of well-being, but the effect is usually modest and transient. Loss of body weight and drenching night sweats profoundly affect quality of life.38 The clinical relevance of constitutional symptoms is underlined by their inclusion as an adverse prognostic risk factor in the IPSS. A specific instrument, the Myeloproliferative Neoplasm Symptom Assessment Form, has been developed to quantitatively assess symptomatic burden, and represents a major advance for rigorously addressing the impact of these manifestations on the patients' quality of life and for objectively measuring the response to conventional and novel therapies.39
A unique case: early/prefibrotic MF
Notwithstanding recent findings indicating reduced overall survival and leukemia-free survival in early/prefibrotic MF compared with typical (“true”) ET, these subjects should be managed as they were “true” ET. They should receive treatment only if they are at high risk for thrombosis. Risk factors include age > 60 years and/or a history of previous major cardiovascular events. Anagrelide should be prudentially avoided because of the possibility of further increase of reticulin.5 Practitioners should be very cautious in discussing with the patient the implications of the term “prefibrotic MF,” considering that this terminology may induce excessive and unnecessary anxiety. Evidence that IFN-α induced clinical benefit/stability of the disease in a significant proportion of MF patients, with documentation of improved BM morphology in some, has been reported recently40 and warrants further evaluation, but at present it cannot be recommended as routine practice.
Promises and pitfalls of novel therapies
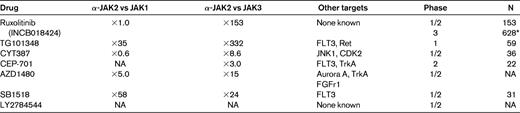
Discovery of the crucial role played by the autonomously activated JAK/STAT signaling pathway in the pathogenesis of MPNs spurred the development of clinical trials using small ATP-competitive molecules (Table 4). Although these are collectively called “JAK2 inhibitors,” all have overlapping activity against other members of the JAK family (including JAK-1, JAK-2, JAK-3, and Tyk2) and possibly other tyrosine kinases as well, and none is specific for mutated JAK2. Indeed, the differences in toxicity profile and efficacy that have been reported in clinical trials may be attributed to some variability in potency, pharmacokinetics, and target selectivity. In addition to the JAK2 inhibitors, other molecules have produced encouraging results in clinical trials and/or preclinical models (Table 5). A few of these, pomalidomide, RAD001 (Everolimus), and inhibitors of histone deacethylases (HDAC), are discussed briefly below.

Pomalidomide
Pomalidomide, an immunomodulating agent with better anticancer and cytokine-regulating activities and lower toxicity compared with thalidomide and lenalidomide, appears to be particularly promising for the treatment of MF-associated anemia. In a phase 2 randomized double-blind trial, 25% of 84 MF patients with anemia responded to pomalidomide alone or in association with prednisone and maintained the response for > 1 year; myelosuppression and neuropathy were infrequent and mild.44 Efficacy of the drug was confirmed in a subsequent study using low-dose pomalidomide (0.5 mg/d) as a single agent, with 38% responses in patients with JAK2V617F-positive disease and a palpable spleen < 10 cm. Intriguingly, the drug was almost ineffective in JAK2 wild-type subjects. Effect on splenomegaly was modest if at all.45 An ongoing phase 3, multicenter, double-blind, placebo-controlled study in transfusion-dependent MF patients is expected to randomize 210 subjects in a 2:1 ratio to the active arm (http://ClinicalTrials.gov identifier NCT01178281).
RAD001 (Everolimus)
Enhanced activation of the mTOR/AKT pathway occurs in MPN cells; RAD001, an inhibitor of mTOR, efficiently prevented the proliferation of JAK2V617F mutated cells in vitro.46 The clinical efficacy of RAD001 has been demonstrated in a phase 1/2 study in 39 subjects with PMF or PPV/PET-MF. Half of the subjects showed improvement of splenomegaly and 60% and 80%, respectively, had complete resolution of systemic symptoms and pruritus. Responses in anemia, including occasional transfusion independence, were recorded in 25% of the patients. Therapy was well tolerated, with modest myelotoxicity and low-grade nonhematological toxicity. This study provided proof-of-concept of the relevance of targeting the mTOR pathway in the therapeutic armamentarium of MF.46
Epigenetic drugs
The contribution of epigenetic alterations to the pathogenesis of MF is supported by the results of recent studies.47 Furthermore, HDAC inhibitors were shown to down-regulate JAK/STAT signaling, probably through additional mechanisms that are largely independent of epigenetic gene regulation. These observations prompted interest in assessing the clinical efficacy of epigenetic drugs.48 However, the hypomethylating agents azacitidine and decitabine did not produce discernible responses in small trials,49,50 and the HDAC inhibitor ITF2357 (givinostat) was minimally effective in MF (unlike in most patients with PV).51 Studies with the HDAC inhibitor LBH589 (panobinostat) in MF are ongoing, and preliminary results are of interest.52
JAK2 inhibitors
Sufficient clinical information is available for 5 of the JAK2 inhibitors under development (Table 4): ruxolitinib (INCB018424), TG101348, lestaurtinib (CEP-701), CYT387, and SB-1515. All of these have been used in PMF or PPV/PET-MF patients with intermediate- or high-risk disease.
Ruxolitinib, a JAK1 and JAK2 inhibitor, was first used in a phase 1/2 trial in 153 patients with MF.53 Treatment was well tolerated (nonhematological grade 1/2 toxicity in < 10% of the patients), with dose-limiting toxicity represented by reversible thrombocytopenia. At the most effective and safest daily dose regimen of 15 mg twice daily, half of the patients had a clinical response with a ≥ 50% reduction of splenomegaly and rapid improvement of constitutional symptoms, cachexia, and exercise tolerance. A similar proportion of patients (14%) became transfusion independent or had anemia worsening as an on-target effect of the drug, whereas thrombocytosis and leukocytosis normalized in more than half of the subjects. The effect on the JAK2V617F allele burden was overall modest. Conversely, responses were independent of the JAK2 mutational status and were no different between PMF and PPV/PET-MF patients. A dramatic normalization of inflammatory cytokines was documented and was correlated with symptomatic improvement; this was most likely attributable to the anti-JAK1 activity. Two phase 3 multicenter studies have now been completed, and preliminary results have been reported recently. The first study, COMFORT-I, assessed the activity of ruxolitinib at 15 or 20 mg twice daily (depending on the baseline platelet level) against placebo in 309 patients.54 The second, COMFORT-II, compared the activity of ruxolitininb against “best-available therapy” in 219 patients randomized 2:1 to the active arm.34 The primary study end point of ≥ 35% spleen volume reduction from baseline as measured by magnetic resonance imaging or computed tomography scan, at 24 (COMFORT-I) or 48 (COMFORT-II) weeks was reached by 41.9% and 28.5% of the patients, respectively, compared with 0.7% and 0% in the control arms (P < .0001). Symptomatic improvement occurred in the majority of patients compared with no change or worsening in the control arm. Toxicity was mainly represented by dose-dependent worsening of anemia; thrombocytopenia did not differ substantially compared with best-available therapy.
TG101348 has greater activity against JAK2 compared with JAK1 (Table 4). Clinical efficacy was evaluated in a phase 1/2 study of 59 patients.55 Dose-limiting toxicity was a reversible and asymptomatic increase in serum amylase level. Usually moderate and transient gastrointestinal adverse events, likely due to the anti-Flt3 activity, occurred in almost 70% of the patients. Worsening of anemia, thrombocytopenia, and neutropenia occurred in 35%, 24%, and 10% of patients, respectively. Most patients with leukocytosis or thrombocytosis had normalization of blood cell counts, and at 6 months almost 60% had attained a ≥ 50% decrease of palpable spleen. Responses were independent of the JAK2V617F mutational status; a ≥ 50% decrease in allele burden was measured in 40% of patients. Symptomatic response was achieved in 50%-75% of patients. Improvement in constitutional symptoms was not associated with measurable changes in inflammatory plasma cytokine levels.
CEP-701 was administered to 22 MF patients, of whom 6 (27%) had clinical improvement. However, gastrointestinal toxicity was remarkable, with diarrhea (72%), nausea (50%), and vomiting (27%). No effect on JAK2V617F allele burden or inflammatory cytokine levels was documented.56 Preliminary results of a phase 1/2 trial suggest an unique activity of CYT387 against anemia.57 Of the 22 subjects evaluable for anemia, 63% qualified for a response in anemia (according to IWG-MRT criteria), with 5 subjects becoming transfusion independent. A > 50% decrease in palpable spleen was documented in 37% of patients. It will be interesting to understand the mechanism(s) by which CYT387 improves anemia. Preliminary information is available for SB1518, which has been used in a phase 2 study of 33 MF patients. The drug was active against splenomegaly and symptoms and, interestingly, did not cause significant hematological toxicity. Diarrhea (grade 3 in 6%) and nausea were reported in 81% and 41% of the patients, respectively.58
It is clearly too early to make firm conclusions about the position of JAK2 inhibitors in the therapeutic scenario of MF, but some considerations apply nonetheless. The first is that because none of these drugs is specific for mutated JAK2, the ultimate effects of treatment reflect the sum of activities also exerted against wild-type JAK2, other members of the JAK family (ie, JAK1), and eventually other still uncharacterized tyrosine kinase targets. Therefore, an optimal ratio between clinical activity and toxicity will be key for choosing among the different JAK2 inhibitors. In addition, preliminary suggestions of a unique mode of action of one inhibitor versus the others are appealing, but need confirmation. Second, because of the essential role that JAK2 plays in normal hematopoiesis and, conversely, the lack of selectivity for the mutated protein, available inhibitors are unlikely to produce molecular remission at doses that are not too myelosuppressive. However, it could be that prolonged administration finally results in a progressive decline of the burden of mutated cells. Finally, although these drugs are quite well tolerated (with some exceptions, in particular relating to gastrointestinal tolerance) information about long-term extrahematologic toxicity are warranted, especially as concerns infectious safety due to the role of JAK family members in immune-mediated responses. All this said, the rapidity and magnitude of clinical improvement that JAK2 inhibitors have produced in cohorts of advanced patients enrolled in clinical trials are indeed remarkable. These have not been seen commonly in clinical practice with any of the “conventional” treatments and largely meet the goals of “symptom-orientated palliation and (improvement of) quality of life” underlined by the ELN experts.20 Therefore, JAK2 inhibitors deserve a major consideration in the symptomatic management of MF.
Conclusions
Excitement that the discovery of the JAK2V617F mutation and the introduction of JAK2 inhibitors in clinical trials could translate into a cure for MF, mimicking the extraordinary success of imatinib in chronic myelogenous leukemia, has not materialized yet. This notwithstanding, our capacity to affect the most compelling clinical needs of patients with MF has undoubtedly improved to the point that there is little question that JAK2 inhibitors will find a definite position in the treatment of selected categories of patients with large, symptomatic splenomegaly and/or overwhelming manifestations of the illness, regardless of their risk category. However, it is important to learn from the large number of preclinical and clinical studies published in the last few years, and to think of MF as a multitarget/multidrug affair, taking into an account the variety and complexity of underlying genetic and cellular abnormalities. Next steps will certainly involve combination therapies.
Disclosures
Conflict-of-interest disclosure: The author has consulted for Novartis. Off-label drug use: Danazol, Cladibrine, Thalidomide, Lenalidomide, RAD001, Azacitabine, Decitabine.
Correspondence
Alessandro M. Vannucchi, Section of Hematology, Department of Critical Care, University of Florence, Viale Morgagni 85, 50134 Florence, Italy; Phone/Fax: 39-055-7947-688; e-mail: amvannucchi@unifi.it.
