
Abstract
According to the World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues, myelodysplastic/myeloproliferative neoplasms are clonal myeloid neoplasms that have some clinical, laboratory, or morphologic findings that support a diagnosis of myelodysplastic syndrome, and other findings that are more consistent with myeloproliferative neoplasms. These disorders include chronic myelomonocytic leukemia, atypical chronic myeloid leukemia (BCR-ABL1 negative), juvenile myelomonocytic leukemia, and myelodysplastic/myeloproliferative neoplasms, unclassifiable. The best characterized of these latter unclassifiable conditions is the provisional entity defined as refractory anemia with ring sideroblasts associated with marked thrombocytosis. This article focuses on myelodysplastic/myeloproliferative neoplasms of adulthood, with particular emphasis on chronic myelomonocytic leukemia and refractory anemia with ring sideroblasts associated with marked thrombocytosis. Recent studies have partly clarified the molecular basis of these disorders, laying the groundwork for the development of molecular diagnostic and prognostic tools. It is hoped that these advances will soon translate into improved therapeutic approaches.
Introduction
The World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues1 includes 5 subgroups of myeloid neoplasms: (1) myeloproliferative neoplasms (MPNs); (2) myeloid/lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1; (3) myelodysplastic syndrome (MDS); (4) myelodysplastic/myeloproliferative neoplasms (MDS/MPNs); and (5) acute myeloid leukemia (AML).
According to Vardiman et al,2 MDS/MPNs are “clonal myeloid neoplasms that at the time of initial presentation have some clinical, laboratory or morphologic findings that support a diagnosis of myelodysplastic syndrome (MDS), and other findings more consistent with myeloproliferative neoplasm (MPN).” These disorders comprise chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML; BCR-ABL1 negative), juvenile myelomonocytic leukemia (JMML), and myelodysplastic/MPNs, unclassifiable (MDS/MPN, U). The best characterized of these latter unclassifiable conditions is the provisional entity defined as refractory anemia with ringed sideroblasts (RARS) associated with marked thrombocytosis (RARS-T).
The diagnosis and treatment of JMML, a disorder of childhood, have been examined in an ad hoc education session at the 2010 American Society of Hematology meeting, and the reader is therefore referred to the related comprehensive article.3 This manuscript focuses on the MDS/MPNs of adulthood: CMML, aCML, and RARS-T.
CMML
CMML is a rare disorder with an estimated incidence of < 1 case per 100 000 persons per year.4 Median age at presentation is ∼ 70 years, and presenting manifestations may include those of BM failure and systemic symptoms. Hepatomegaly and splenomegaly are found in some patients, and the WBC count is typically increased.
WHO diagnostic criteria for CMML
The 2008 WHO diagnostic criteria for CMML are as follows (see also previous paper by Dr Vardiman5 ):
Persistent peripheral blood monocytosis (> 1 × 109/L);
No Philadelphia chromosome or BCR-ABL1 fusion gene;
No arrangement of PDGFRA or PDGFRB (these rearrangements should be specifically excluded in cases with eosinophilia);
Less than 20% blasts in the peripheral blood and the BM (blasts includes myeloblasts, monoblasts, and promonocytes); and
At least one of the following: (a) dysplasia in one or more cell lines, (b) an acquired clonal cytogenetic abnormality or molecular genetic abnormality present in hematopoietic cells, or (c) persistence of monocytosis for at least 3 months and no evidence of other causes of monocytosis (such infection, inflammation. or malignancy).
Based on blast counts, CMML is further subdivided into:
CMML-1: blasts (including promonocytes) < 5% in the peripheral blood and < 10% in the BM;
CMML-2: blasts from 5%-19% in the peripheral blood and from 10%-19% in the BM or when Auer rods are present irrespective of blast count.
Based on the WBC count, CMML had been previously separated into myelodysplastic-like (WBC < 13 × 109/L) and myeloproliferative-like CMML (WBC ≥ 13 × 109/L). This distinction is likely arbitrary, but the WBC represents an important prognostic factor (see below).
Representative pictures of peripheral blood and BM smears from CMML patients are shown in Figure 1. According to the WHO criteria, blasts include myeloblasts, monoblasts, and promonocytes, but not abnormal monocytes. However, these latter are common in CMML (Figure 1B), and the distinction between promonocytes and abnormal monocytes may be problematic, although ad hoc morphologic criteria have been proposed.6

Representative peripheral blood and BM smears from patients with CMML. (A) Peripheral blood smear showing monocytosis with morphologically normal monocytes. May-Grünwald-Giemsa (MGG) staining; magnification, ×400. (B) Peripheral blood smear showing monocytes with nuclear and cytoplasmic abnormalities. MGG staining; magnification, ×1250. (C) BM smear showing high cellularity and hyperplasia of the granulocytic series with < 10% blasts (CMML-1). Erythroid cells are morphologically normal. MGG staining; magnification, ×1000. (D) BM smear from a patient with CMML-2 showing hyperplasia of monocytes and their precursors. The sum of blasts plus the promonocytes is 18%. MGG staining; magnification, ×1000.
Representative peripheral blood and BM smears from patients with CMML. (A) Peripheral blood smear showing monocytosis with morphologically normal monocytes. May-Grünwald-Giemsa (MGG) staining; magnification, ×400. (B) Peripheral blood smear showing monocytes with nuclear and cytoplasmic abnormalities. MGG staining; magnification, ×1250. (C) BM smear showing high cellularity and hyperplasia of the granulocytic series with < 10% blasts (CMML-1). Erythroid cells are morphologically normal. MGG staining; magnification, ×1000. (D) BM smear from a patient with CMML-2 showing hyperplasia of monocytes and their precursors. The sum of blasts plus the promonocytes is 18%. MGG staining; magnification, ×1000.
Chromosomal abnormalities, somatic mutations, and their diagnostic and prognostic relevance
Investigators in the Spanish MDS registry recently studied 414 CMML patients who had a successful conventional cytogenetic analysis at diagnosis.7 Cytogenetic abnormalities were found in 110 of 414 (27%) patients. Multivariable analysis of survival and progression to AML allowed 3 cytogenetic risk categories to be identified: (1) low-risk (normal karyotype or loss of Y chromosome as a single anomaly), (2) high-risk (presence of trisomy 8 or abnormalities of chromosome 7 or complex karyotype), and (3) intermediate risk (all other abnormalities). The actuarial median survival of patients within these 3 cytogenetic subgroups was 37, 18, and 11 months, respectively.
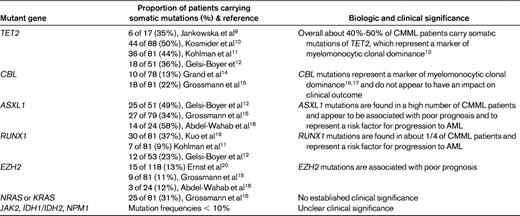
Until recently, the most common known molecular abnormality in CMML was NRAS or KRAS mutations, seen in approximately 1/3 of cases but of uncertain significance with regard to pathogenesis and prognosis.8 In the last few years, however, improved technologies have enabled the identification of novel mutant genes in a significant number of CMML patients, specifically TET2, CBL, ASXL1, RUNX1, and EZH2. As shown in Table 1, TET2 and CBL mutations are mainly a marker of myelomonocytic clonal proliferation, while ASXL1, RUNX1 and EZH2 mutations are associated with disease progression. A causative role of defective TET2 activity in the clonal proliferation of myelomonocytic cells that characterizes CMML is supported by recent findings of Ko et al.21 These investigators showed that TET2 mutations associated with MDS/MPN, including CMML, compromise catalytic activity of the protein, and that depletion of Tet2 in mouse hematopoietic precursors in vitro results in skewed differentiation toward monocyte/macrophage lineages.
In a recent study, Delva et al22 have shown that, in mice, tissue-targeted deletion of the gene encoding transcription intermediary factor 1γ (Tif1γ) leads to development of a CMML-like myeloproliferative disease with monocytic features. Interestingly, the human TIF1G gene expression was almost undetectable in sorted leukemic cells of ∼35% of patients with CMML, and this decreased expression was related to the hypermethylation of CpG islands and a specific pattern of histone modifications on the gene promoter. This suggests that reduced expression of TIF1G may represent a pathogenetic mechanism of human CMML.
Diagnostic work-up
The current diagnostic approach to CMML includes examination of peripheral blood smear and BM aspirate, BM biopsy, and cytogenetic analysis. Additional optional investigations are provided in Table 2.
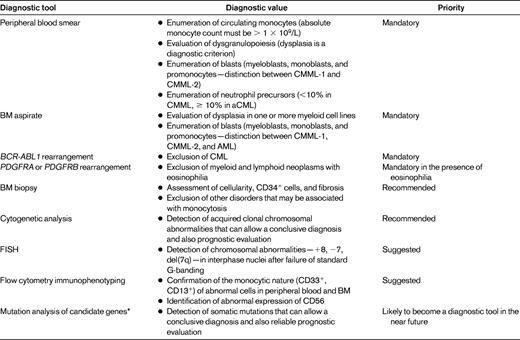
The diagnosis of CMML is straightforward in the presence of a combination of persistent monocytosis and a clonal cytogenetic abnormality or somatic mutation in myeloid cells. Conversely, the absence of a clonal abnormality makes the diagnosis of CMML uncertain, especially in CMML-1 cases, in which BM and peripheral blood counts are low.
Risk assessment
The International Prognostic Scoring System (IPSS) was developed in a patient population that included CMML, and may therefore be used for risk assessment; however, the IPPS was only built and validated for nonproliferative CMML patients with WBC below 12 × 109/L.23 Onida et al24 generated a specific prognostic score based on anemia, presence of circulating immature myeloid cells, absolute lymphocyte count, and percentage of BM blasts. This model identified 4 subgroups of patients with median survival times of 24, 15, 8, and 5 months for low, intermediate-1, intermediate-2, and high risk, respectively. More recently, Kantarjian et al validated a new risk model for MDS and showed that it is also applicable to patients with CMML.25
Germing et al26 analyzed 266 patients with CMML-1 and 73 patients with CMML-2. Median survival time was 20 months for CMML-1 and 15 months for CMML-2, and the risk of progression to AML was higher in the latter patients. As shown in Table 3, Such et al7 identified several prognostic factors independently associated with clinical outcome. The WBC predicted leukemic transformation, with the cumulative probability of AML evolution at 2 and 5 years being 15% and 29%, respectively.
Prognostic factors in CMML*
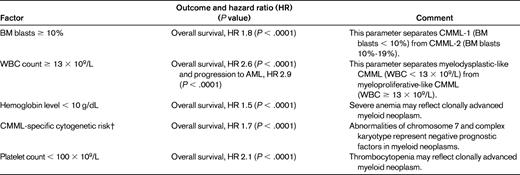
Results of multivariate analysis of survival and risk of AML evolution in the study by Such et al.7
†(1) Low-risk, normal karyotype or loss of Y chromosome as a single anomaly; (2) high-risk, presence of trisomy 8 or abnormalities of chromosome 7 or complex karyotype; and (3) intermediate-risk, all other abnormalities. The actuarial median survival of patients within these 3 cytogenetic risk groups was 37, 18, and 11 months, respectively.
Therapeutic options
Only few clinical trials have been performed in patients with CMML, and most of the studies published so far are retrospective and have selection biases, making it difficult to draw firm conclusions. Therapeutic options for patients include best supportive care, demethylating agents (azacitidine and decitabine), hydroxyurea, and allogeneic stem cell transplantation.
Azacitidine has been approved by the US Food & Drug Administration (FDA) for “treatment of CMML,” and by the European Medicines Agency (EMA) for “treatment of CMML with 10%-29% marrow blasts without myeloproliferative disorder.” The available evidence suggests that azacitidine may be effective in some patients with CMML, especially those with BM blasts ≥ 10%.27,28 An oral formulation of azacitidine has been recently found to be active in patients with CMML.29 Decitabine has been approved by the FDA for the treatment of patients with MDS of all FAB subtypes (including CMML) and intermediate-1, intermediate-2, and high IPSS risk. A recent reevaluation of 3 clinical trials reports an overall response rate of 25% in CMML patients treated with decitabine.30
Hydroxyurea was found to be superior to etoposide in a randomized study in advanced CMML that enrolled patients with elevated WBC count (mean value between 25 and 31 × 109/L).31 Median actuarial survival was 20 months in the hydroxyurea arm versus 9 months in the etoposide arm.
Unfortunately, none of the above drugs is able to substantially modify the natural history of CMML, which is inevitably associated with a poor outcome, with median survival times of ∼ 25-40 months in the low-risk group versus 5-11 months in the high-risk group.7,24 As in MDS, allogeneic stem cell transplantation remains the only curative treatment for CMML. However, most patients are above the age of 70 and the transplantation studies reported so far include highly selected patients, making it difficult to properly assess the impact of this therapeutic option.32 In a retrospective study on 85 CMML patients with follow-up extending to 19 years, progression-free survival was 38% at 10 years, indicating that allogeneic transplantation can indeed cure a significant number of patients.33
Taking into account the limited evidence available, allogeneic stem cell transplantation should be considered in patients up to the age of 65-70 (depending on the specific center's policy and the presence and character of comorbidities) who have a compatible donor. Patients who are not eligible for allogeneic stem cell transplantation should be enrolled in clinical trials aimed at evaluating novel treatments for CMML: a search performed at the beginning of May 2011 at www.ClinicalTrials.gov showed 409 studies involving patients with CMML.
Patients who are not eligible for allogeneic stem cell transplantation nor for clinical trials may benefit from treatment with azacitidine, particularly those with CMML-2 and unfavorable cytogenetics. Cytoreductive therapy with hydroxyurea should instead be considered in patients with elevated WBC count (≥ 13 × 109/L). In anemic patients, regular RBC transfusions remain the mainstay of therapy.
aCML, BCR-ABL1 negative
Compared with CMML, aCML is even more rare, with an estimated incidence of 1 case per 1 000 000 persons per year or less. Presenting clinical features include symptoms of BM failure, systemic symptoms, and splenomegaly.
The 2008 WHO diagnostic criteria for aCML have been previously discussed by Dr. Vardiman.5 In brief, aCML is basically a neutrophilic leukocytosis with dysgranulopoiesis and circulating immature granulocytes, and its diagnosis relies on weak grounds at present. Representative peripheral blood and BM smears are reported in Figure 2. In practice, the differentiation between aCML and the hypercellular phase of primary myelofibrosis may be challenging, even taking into account the diagnostic value of JAK2 and MPL mutations. In addition, the differential diagnosis between aCML and chronic neutrophilic leukemia is difficult, because the only distinctive feature is the proportion of immature neutrophils (≥ 10% in aCML and < 10% in CNL).

Representative peripheral blood and BM smears from patients with aCML, BCR-ABL 1 negative. (A) Peripheral blood smear showing leukocytosis. On the upper left is a metamyelocyte; lower left, a dysplastic neutrophil. May-Grünwald-Giemsa (MGG) staining; magnification, × 400. (B) Peripheral blood smear. A neutrophil with nuclear hypersegmentation (center) and a red cell with a Howell-Jolly body (top); left, polychromatic red cells and giant platelets. MGG staining; magnification, × 1250. (C) BM smear showing granuloblastic hyperplasia and erythroid hypoplasia. Mature neutrophils are agranular and show abnormal nuclear segmentation. Note the micromegakaryocyte (center). MGG staining; magnification, × 640. (D) Syndrome of abnormal chromatin clumping. Peripheral blood smear showing leukocytosis, immature granulocytes, and neutrophils with abnormal condensation of the nuclear chromatin. MGG staining; magnification, × 1250.
Representative peripheral blood and BM smears from patients with aCML, BCR-ABL 1 negative. (A) Peripheral blood smear showing leukocytosis. On the upper left is a metamyelocyte; lower left, a dysplastic neutrophil. May-Grünwald-Giemsa (MGG) staining; magnification, × 400. (B) Peripheral blood smear. A neutrophil with nuclear hypersegmentation (center) and a red cell with a Howell-Jolly body (top); left, polychromatic red cells and giant platelets. MGG staining; magnification, × 1250. (C) BM smear showing granuloblastic hyperplasia and erythroid hypoplasia. Mature neutrophils are agranular and show abnormal nuclear segmentation. Note the micromegakaryocyte (center). MGG staining; magnification, × 640. (D) Syndrome of abnormal chromatin clumping. Peripheral blood smear showing leukocytosis, immature granulocytes, and neutrophils with abnormal condensation of the nuclear chromatin. MGG staining; magnification, × 1250.
Regarding somatic mutations, TET2 mutations are found in approximately one-third of aCML patients,8 and CBL mutations in ∼ 8%.14 Ernst et al20 reported that 9 of 70 (13%) aCML cases have EZH2 mutations, confirming the considerable overlap between aCML and CMML at the molecular level as well. A significant step forward in our comprehension of the molecular basis of aCML would be to identify the mutant gene(s) responsible for the release of immature myeloid cells in the peripheral blood, a typical feature of aCML not found in CMML.
In a retrospective study conducted at the M.D. Anderson Cancer Center,34 the median overall survival was 24 months. Multivariable analysis identified older age (> 65 years), anemia (hemoglobin < 10 g/dL), and severe leukocytosis (WBC > 50 × 109/L) as variables with independent prognostic significance for poor survival. A more recent Italian study essentially confirmed these findings.35 Age > 65 years and elevated WBC count (≥ 50 × 109/L) were independently associated with reduced survival, and transfusion dependency and splenomegaly predicted for leukemic transformation.
A peculiar variant of aCML is the syndrome of abnormal chromatin clumping in leucocytes,36 in which neutrophils and immature RBCs exhibit exaggerated clumping of their nuclear chromatin (Figure 2D). The clinical course of these patients is similar to that of other patients with aCML.
Another interesting variant of aCML is the MDS/MPN associated with the t(8;9)(p22; p24) translocation that fuses JAK2 to PCM1.37,38 Although some cases present with eosinophilia and may be classified as chronic eosinophilic leukemia, approximately half of the patients have a clinical picture of aCML.39 The PCM1-JAK2-fusion is likely to be a potential target of JAK2 inhibitors. The involvement of ETV6-JAK2 fusions has also been described in the development of aCML.40
Overall, aCML is generally associated with poor outcome, and no drug has so far proved to be effective in this disorder. Therefore, patients who have a donor and are eligible for the procedure should be considered for allogeneic stem cell transplantation, and the remaining ones should be enrolled in clinical trials. Hydroxyurea can be used for controlling excessive leukocytosis and/or symptomatic splenomegaly.
Refractory anemia with ring sideroblasts associated with marked thrombocytosis
The sideroblastic anemias are a heterogeneous group of inherited and acquired disorders characterized by anemia of varying severity and the presence of ring sideroblasts in the BM.41,42 Ring sideroblasts are erythroblasts with iron-loaded mitochondria that are visualized by Prussian blue staining as a perinuclear ring of blue granules.43 Most of the iron deposited in these perinuclear mitochondria of ring sideroblasts is present in the form of mitochondrial ferritin.44 The presence of ring sideroblasts in the BM (15% or more) is a marker of the MDS defined as RARS, which is associated with a benign clinical course.45
WHO diagnostic criteria for RARS-T
The 2008 WHO diagnostic criteria for RARS-T are as follows:
Refractory anemia associated with erythroid dysplasia and ringed sideroblasts 15% or greater;
Less than 5% blasts in the BM;
Platelet count 450 × 109/L or greater;
Presence of large atypical megakaryocytes similar to those observed in BCR/ABL1-negative MPN; and
Absence of del(5q), t(3;3)(q21;q26), or inv(3)(q21q26).
Because thrombocytosis is a simple reliable parameter, and ringed sideroblasts represent a robust morphological abnormality, the diagnosis of RARS-T is relatively easy. These disorders must be primarily distinguished from essential thrombocythemia (ET), and this distinction is easily done through a Perls staining of a BM smear. Representative peripheral blood and BM smears from patients with RARS-T are shown in Figure 3.

Representative peripheral blood and BM smears from patients with refractory anemia with ring sideroblasts and marked thrombocytosis. (A) Peripheral blood smear showing thrombocytosis. May-Grünwald-Giemsa (MGG) staining; magnification, × 1000. (B) BM smear showing numerous clustered megakaryocytes that are markedly enlarged and similar to those observed in essential thrombocythemia. MGG staining; magnification, × 500. (C) BM smear showing erythroid hyperplasia with macro-megaloblastoid changes. MGG staining; magnification, × 1000. (D) BM smear after Perls staining showing numerous ring sideroblasts. Magnification, × 1000.
Representative peripheral blood and BM smears from patients with refractory anemia with ring sideroblasts and marked thrombocytosis. (A) Peripheral blood smear showing thrombocytosis. May-Grünwald-Giemsa (MGG) staining; magnification, × 1000. (B) BM smear showing numerous clustered megakaryocytes that are markedly enlarged and similar to those observed in essential thrombocythemia. MGG staining; magnification, × 500. (C) BM smear showing erythroid hyperplasia with macro-megaloblastoid changes. MGG staining; magnification, × 1000. (D) BM smear after Perls staining showing numerous ring sideroblasts. Magnification, × 1000.
Molecular basis of RARS and RARS-T
CD34+ cells from patients RARS have a particular gene-expression profile characterized by up-regulation of mitochondrial-related genes, in particular those of heme synthesis (eg, ALAS2),46 and down-regulation of ABCB7, a gene encoding a protein involved in the transport of iron/sulfur clusters from the mitochondria to the cytoplasm.47 RARS is characterized by the accumulation of iron in mitochondria and by the overexpression of mitochondrial ferritin, which is encoded by the FTMT gene.42,44,48
The molecular basis of RARS has been very recently deciphered in a study based on the use of massively parallel sequencing technology to identify somatically acquired point mutations across all protein-coding exons.49 Recurrent somatically acquired mutations were identified in SF3B1, a gene encoding an RNA-splicing factor, implicating abnormalities of mRNA splicing,
With respect to RARS-T, we have also found that these patients consistently showed up-regulation of ALAS2 and down-regulation of ABCB7 in CD34+ cells, but several other genes were differentially expressed, including PSIP1 (LEDGF), CXCR4, and CDC2L5.50 More importantly, 11 of 19 (58%) patients with RARS-T carried JAK2 or MPL mutations in circulating granulocytes, whereas these somatic mutations were not detected in any of the RARS patients. These results were confirmed by Flach et al, who detected the JAK2 (V617) mutation in 15 of 19 (79%) patients with RARS-T.51 In contrast, none of the 19 patients analyzed carried CBL mutations. Interestingly, somatic mutations of TET2 were detected in 5 of 19 (26%) patients, of which 3 of 5 also carried JAK2 (V617F).
These observations suggest that RARS-T is indeed a myeloid neoplasm with both myelodysplastic and myeloproliferative features at the molecular and clinical level, and that it may develop from RARS through the acquisition of somatic mutations of JAK2, MPL, or other as-yet-unknown genes.50,52 A schematic representation of the multistep molecular pathogenesis of RARS-T based on the available evidence49,53 and currently unpublished data are reported in Figure 4.

Schematic representation of the multistep molecular pathogenesis of RARS-T supporting its nature of true MDS/MPN (combination of SF3B1 and JAK2 or MPL mutations). The occurrence of a somatic mutation of SF3B1 causes mitochondrial iron overload, ineffective erythropoiesis, and anemia, typical myelodysplastic features of RARS. The subsequent occurrence of a somatic mutation of JAK2 or MPL involves the acquisition of myeloproliferative features, including thrombocytosis. This hypothetical model is based on data still unpublished at the time of this writing.
Schematic representation of the multistep molecular pathogenesis of RARS-T supporting its nature of true MDS/MPN (combination of SF3B1 and JAK2 or MPL mutations). The occurrence of a somatic mutation of SF3B1 causes mitochondrial iron overload, ineffective erythropoiesis, and anemia, typical myelodysplastic features of RARS. The subsequent occurrence of a somatic mutation of JAK2 or MPL involves the acquisition of myeloproliferative features, including thrombocytosis. This hypothetical model is based on data still unpublished at the time of this writing.
Clinical course of RARS-T and treatment
In a retrospective study,54 RARS-T patients were found to have a median overall survival of 71 months, comparable with RARS but less favorable than ET. A European study has recently confirmed that the clinical course of RARS-T is more similar to that of RARS than to that of ET.55 This underlines the importance of distinguishing between RARS-T and ET in patients with borderline values of hemoglobin.
There is no evidence that cytoreductive therapy is useful in patients with RARS-T, and the administration of hydroxyurea might result in a worsening of anemia. Huls et al recently treated 2 patients with RARS-T who were JAK2 (V617F)–positive and transfusion dependent with lenalidomide.56 Both patients became transfusion independent and one achieved a complete molecular remission.
Conclusions and perspectives
Recent studies have partly clarified the molecular basis of MDS/MPN, in particular CMML and RARS-T, creating the basis for the development of molecular diagnostic and prognostic tools. Unfortunately, these achievements have not yet translated into novel therapeutic approaches and it is therefore of fundamental importance to enroll patients with MDS/MPN in prospective clinical trials.
Note added in proof:
Yoshida et al57 have recently reported a study of whole-exome sequencing of myelodysplasia specimens, which unexpectedly revealed novel pathway mutations involving multiple components of the RNA splicing machinery. With respect to CMML, targeted resequencing showed somatic mutations in genes encoding key components of the spliceosome (SRSF2, USAF35, ZRSR2, SF3A1, U2AF65, and SF3A1) in 54.5% of patients. This clearly indicates that abnormalities of splicing machinery play a major role in the pathophysiology of CMML, being likely responsible for its myelodysplastic component.
Acknowledgments
Studies on MDS/MPNs performed at the Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy, are supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC, Milan), Fondazione Cariplo (Milan), and Regione Lombardia (Milan) to M.C.
Disclosures
Conflict-of-interest disclosures: The authors declare no competing financial interests. Off-label drug use: Lenalidomide for the treatment of refractory anemia with ring sideroblasts and marked thrombocytosis.
Correspondence
Mario Cazzola, MD, Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, 27100 Pavia, Italy; Phone: +39-0382-503595; Fax: +39-0382-502250; e-mail: mario.cazzola@unipv.it.
