
Abstract
In the past decade, a series of technological advances have revolutionized our ability to interrogate cancer genomes, culminating in whole-genome sequencing, which provides genome-wide coverage at a single base-pair resolution. As sequencing technologies improve and costs decrease, it is likely that whole-genome sequencing of cancer cells will become commonplace in the diagnostic workup of patients with acute myelogenous leukemia (AML) and other cancers. The unprecedented molecular characterization provided by whole-genome sequencing offers the potential for an individualized approach to treatment in AML, bringing us one step closer to personalized medicine. In this chapter, we discuss how next-generation sequencing is being used to study cancer genomes. Recent publications of whole-genome sequencing in AML are reviewed and current limitations of whole-genome sequencing are examined, as well as current and potential future clinical applications of whole-genome sequencing.
Introduction
Since Nowell and Hungerford identified the t(9;22) translocation associated with chronic myeloid leukemia,1 a wealth of data has accumulated showing that the karyotype and mutation status of certain genes provide important prognostic, and in some cases, therapeutic information for acute myelogenous leukemia (AML). However, standard cytogenetics has a limited resolution (3-5 megabases) to detect chromosomal abnormalities and genotyping of individual genes is expensive; therefore, as the number of mutations identified in AML increases, this method may soon become impractical. During the last decade, the technology to interrogate cancer genomes has rapidly advanced. The resolution for variant detection was improved first with array-based technologies using comparative genome hybridization (CGH) and single nucleotide polymorphism (SNP) analysis, and now with whole-genome sequencing, which provides unbiased, genome-wide coverage at a single base-pair resolution. Gene-expression profiling and CGH/SNP analysis of AML have provided important insights into AML classification and biology, and this has been recently reviewed elsewhere.2–4 This chapter will focus on the use of next-generation sequencing technologies to characterize AML genomes.
In the past, the size and complexity of the human genome (3 billion base pairs) made the sequencing of human cancer genomes impractical. Two major advances helped to overcome these obstacles. First, the generation of the draft sequence of the human genome by the Human Genome Project in 2001 provided a road map of the human genome.5 Second, technological advances in DNA sequencing dramatically reduced the cost and time required to sequence genomes (Figure 1). Whereas the Human Genome Project took more than 10 years and several billion dollars to sequence the first human genome, current estimates are 6 weeks and $20 000 per human genome ($40 000 for paired tumor/normal genomes). Therefore, we are rapidly approaching the time when sequencing the genomes of patients with cancer will be practical in the clinical setting.

Timeline for sequencing of AML genomes. Cost and time estimates are for paired leukemia/normal genomes and include data production, bioinformatic analysis, validation, and interpretation. Current cost and time estimates to sequence a human genome were provided by Dr Richard Wilson, Director, Genome Institute at Washington University (St Louis, MO).
Timeline for sequencing of AML genomes. Cost and time estimates are for paired leukemia/normal genomes and include data production, bioinformatic analysis, validation, and interpretation. Current cost and time estimates to sequence a human genome were provided by Dr Richard Wilson, Director, Genome Institute at Washington University (St Louis, MO).
Next-generation sequencing of cancer
The development of massively parallel sequencing (termed next-generation or second-generation sequencing) revolutionized our ability to analyze cancer genomes. Interested readers are referred to several recent reviews describing next-generation sequencing technologies.6,7 In brief, massively parallel sequencing results in the generation of millions of short (50-100 nucleotides) DNA sequences simultaneously. These sequences are then mapped back to the human reference genome to generate a picture of the cancer genome. For studies of cancer, it is key to sequence both the tumor and the normal tissue (eg, skin tissue) from the individual. There are 3-4 million inherited sequence variants per human genome (and hundreds of copy number variants). Consequently, the majority of sequence variants identified in a cancer genome are inherited polymorphisms and not acquired mutations. Therefore, a comparison of a tumor genome with its paired normal genome is required to efficiently identify acquired (somatic) sequence variants.
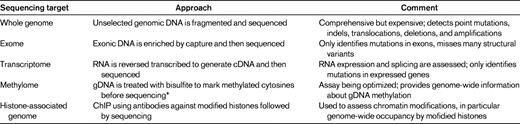
Currently, there are several different ways in which next-generation sequencing is being applied to study cancer genomes. The goals, advantages, and limitations of each approach are summarized below and in Table 1. Ultimately, combinations of approaches (eg, whole-genome and transcriptome sequencing) may be required to comprehensively study cancer cells.
Whole-genome sequencing
The goal of whole-genome sequencing is to sequence the entire genome. Typically, this requires at least 30- to 40-fold haploid coverage of the genome (∼ 100 Gbp of sequence) to achieve adequate diploid coverage for comprehensive mutation discovery. The advantages of whole-genome sequencing include: (1) the entire genome is surveyed, not just coding genes, and (2) structural variants, including deletions, amplifications, chromosomal translocations, and uniparenteral disomy, are readily identified. The major limitations are cost and the complexity of the data analysis. As cost and bioinformatic approaches to analyzing sequence data advance, whole-genome sequencing is likely to become the dominant platform for mutation detection.
Exome sequencing
The goal of exome sequencing is to selectively sequence the 1%-2% of the genome containing coding genes, microRNAs, and other noncoding RNAs. In brief, genomic DNA is hybridized to beads (or arrays) containing probes designed to capture the approximately 200 000 exons in the human genome. The DNA “captured” on the beads is then subjected to next-generation sequencing. The major advantages of exome sequencing are reduced cost and relatively deep sequence coverage because only 1%-2% of the genome is analyzed. However, exome sequencing will not detect mutations in regions outside of the exome (> 98% of the genome) and will not detect most structural variants, such as chromosomal translocations with intronic break points.
Transcriptome sequencing
The goal of transcriptome sequencing is to sequence all transcribed genes,8 including both coding and noncoding RNAs. The advantages of transcriptome sequencing include: (1) quantitative information about gene-expression levels are obtained, (2) posttranscriptional changes in gene expression such as alternative splicing are detected, and (3) fusion transcripts produced by chromosomal rearrangements can be detected. Similar to sequencing the exome, transcriptome sequencing does not detect mutations in noncoding regions of the genome. It cannot detect mutations that cause the loss of one or both copies of a gene or mutations that accelerate RNA turnover (eg, frameshift or nonsense mutations that cause “nonsense mediated decay”). Moreover, transcriptome sequencing is biased toward abundantly expressed transcripts and therefore the sequencing coverage of genes expressed at lower levels can be low or absent.
Other sequencing applications
There are emerging next-generation sequencing applications to characterize genome-wide epigenetic modifications in cancer cells. This is particularly relevant to the study of AML, because genes regulating DNA methylation are frequently mutated in AML, including DNMT3A. Next-generation sequencing techniques have been developed to map genome-wide DNA methylation at a single-base resolution9 and to assess chromatin structure by mapping the location of posttranslationally modified (eg, methylated and acetylated) histones on the genome.10
Identification of novel somatic mutations in AML
Traditional approaches to mutation discovery in cancer rely on sequencing of a preselected group of genes. Typically, genes or gene family members already implicated in cancer are sequenced. This directed sequencing approach (in effect “looking under the lamppost”), while yielding important insights into cancer biology, is inherently limited. In contrast, genomic approaches study the entire cancer genome and thus provide a nonbiased way to look for mutations. Moreover, by simultaneously evaluating all genes, whole-genome sequencing is an ideal platform to assess cooperativity between gene mutations.
In a landmark study published in 2008, Ley et al reported the sequence of the first primary cancer genome, a case of FAB M1-AML.11 Since then, the sequence of 3 additional AML genomes have been reported: an additional case of M1-AML (normal karyotype)11–13 and 1 case each of M3-AML14 and therapy-related AML (complex karyotype).15 In addition, exome sequencing was reported in 14 cases of M5-AML.16
These studies have begun to define several important global features of AML. First, there are only a relatively small number of somatically acquired single nucleotide variants (SNVs or “point mutations”) in AML cells. Within coding regions, the number of somatic SNVs ranged from 10-26 in the 4 sequenced AML genomes.11–15 This suggests that AML does not result from genomic instability, but rather may be associated with a small number of oncogenic “driver” mutations. Second, most acquired AML-associated SNVs are private: of the 59 reported SNVs in the coding regions of these 4 genomes, no gene was mutated in 2 or more patients. This suggests that many of these mutations are likely to be personal “passenger” mutations, which are random and do not contribute to AML pathogenesis.
Despite the lack of recurring mutations within these 4 genomes, these studies identified 2 novel AML-associated genes that were found to be frequently mutated in AML and contribute to leukemogenesis. Mardis et al identified mutations in IDH1 in the second AML genome published and found an additional 16 patients carrying IDH1 mutations.13 Mutations in IDH1 and IDH2 have since been identified in AML patients in several studies, occur at a frequency of 7%-16%, and appear to be mutually exclusive of one another and of acquired TET2 mutations.17,18 A recent study found that mutations in IDH1 or IDH2 disrupted TET2 function and led to a hypermethylation phenotype with impaired hematopoietic differentiation,19 although a second study found impaired methylation associated with TET2 mutations.20 Efforts are ongoing to develop targeted therapeutics that might abrogate these effects.
Acquired mutations in DNMT3A, a de novo DNA methyltransferase, were identified by next-generation sequencing of AML samples by 3 independent groups. Yamashita et al used targeted exome sequencing to identify acquired mutations in DNMT3A at amino acid R882 in 3 of 71 (4.2%) AML cases.21 Ley et al identified a single base-pair deletion in DNMT3A by whole-genome sequencing and identified acquired mutations (including deletions, frame-shifts, nonsense, and missense mutations) in an additional 62 of 281 AML patients (22.1%).12 They showed that DNMT3A mutations were more common in AML with normal cytogenetics (33.7%) or M5-AML cytomorphology (57.1%) and were associated with an adverse outcome. Finally, Yan et al used exome sequencing to identify missense mutations at R882 and at 3 other amino acid positions in 23 of 112 (20.5%) patients with AML-M5.16 It is currently unclear whether some of these mutations (eg, the highly recurrent R882 mutations) might be associated with a gain of function. However, Challen et al found that Dnmt3a-deficient HSCs expanded significantly after serial transplantation, suggesting that the loss of DNMT3A could contribute directly to leukemogenesis.22 A detailed description of the clinical utility of IDH1, IDH2, and DNMT3A mutations in the risk stratification and treatment of AML is provided in the companion chapter by Döhner and Gaidizik.
Identification of cancer susceptibility genes in AML
Cancer susceptibility is an important consideration in individuals presenting with cancers at an early age, with multiple primary cancers, or with a suggestive family history. Determining the genetic basis for cancer susceptibility is desirable because it may affect the prevention, early detection, and treatment of associated neoplasms. There are more than 100 genes implicated in cancer susceptibility.23 Genetic testing for all of these genes is cost-prohibitive, impractical, and frequently unrevealing, because many of the genes contributing to cancer susceptibility have not yet been identified. Whole-genome sequencing provides a comprehensive, unbiased approach to identify mutations in genes contributing to cancer susceptibility.
We recently reported the results of whole-genome sequencing in a woman who presented at age 37 with breast cancer, at age 39 with ovarian cancer, and at age 42 with rapidly fatal therapy-related AML.15 Although her family history of cancer was unremarkable, the early onset of multiple cancers suggested a cancer susceptibility syndrome. Commercial genetic testing for mutations in BRCA1 and BRCA2 was negative. Consistent with current guidelines for assessing familial cancer risk, no further genetic testing was performed. Whole-genome sequencing revealed a novel, heterozygous 3-kb deletion removing exons 7-9 of TP53 in the patient's normal skin DNA, which was homozygous in the leukemia DNA as a result of uniparental disomy. Genetic testing of the patient's mother and the absence of a history of cancer in the patient's extended family suggested that this mutation arose spontaneously in the patient. The finding of the germline TP53 mutation has important clinical implications for the patient's children. Carriers of a deleterious TP53 germline mutation have an approximately 90% lifetime risk of developing cancer, and current guidelines from the National Comprehensive Cancer Network (NCCN) recommend annual screening for certain cancers.24 Therefore, although whole-genome sequencing did not affect the management of this patient's AML, it did provide clinically important, actionable information for her family.
Characterization of complex genomes and cryptic translocations in AML
Cytogenetic translocations such as t(15;17), t(8;21), and inv(16) have become an essential part of AML diagnostics and are used to predict response to therapy. This is perhaps best illustrated by the use of all trans-retinoic acid (ATRA) therapy in patients with t(15;17) and the associated PML-RARA fusion oncogene. However, standard cytogenetics or FISH analysis do not detect all translocations producing PML-RARA. Cryptic insertions and/or complex translocations that produce functional PML-RARA transcripts have been described.25–27 We recently described a case in which whole-genome sequencing was used clinically to identify an actionable cryptic translocation in a patient with M3-AML.14
Case history
A 39-year-old woman presented with morphologic and clinical features of M3-AML. However, chromosomes 15 and 17 appeared to be normal by metaphase cytogenetics, but multiple chromosomal alterations were identified, including unusual translocations and the loss of whole chromosomes, consistent with high-risk complex cytogenetics. FISH was not diagnostic for PML-RARA. The patient entered complete remission after induction therapy with cytarabine and idarubicin, and her sister was found to be an HLA match. This presented a diagnostic conundrum with divergent treatment options: does she have APL (which should be treated with ATRA consolidation) or does she have complex cytogenetic AML (which should be treated with allogenic stem cell transplantation in first remission)?
To address this issue, whole-genome sequencing was performed. The patient was empirically treated with 2 cycles of arsenic consolidation, which imposed a deadline of 8 weeks to complete the sequencing, perform the bioinformatic analysis, and validate the findings. This deadline was met, and whole-genome sequencing revealed that a 77-kb fragment from the LOXL1-PML locus on chromosome 15 was cleanly inserted into the RARA locus on chromosome 17, creating an in-frame PML-RARA fusion allele. Admittedly, alternative strategies may have identified this insertion. RT-PCR to detect the PML-RARA fusion transcript was not initially performed and clinical samples were not available later for analysis. Other research-based approaches could be envisioned (eg, long-range PCR or nested PCR using a linker-ligated library). However, each of these requires personalized design and iterative trouble-shooting. In summary, this case illustrates that, already in 2011, whole-genome sequencing can be clinically valuable in appropriately selected patients. The diagnostic information obtained from whole-genome sequencing allowed this patient to avoid the hazards and expense of an allogeneic stem cell transplantation in favor of ATRA consolidation.
Current limitations of whole-genome sequencing
Although clearly a valuable and promising approach to analyzing AML (and other cancers), there are some technical limitations of whole-genome sequencing. The ability of next-generation sequencing to detect sequence variants is dependent on the “read-depth,” the number of unique times a single nucleotide is sampled. Whole-genome sequencing typically aims to cover each nucleotide with an average of 30-40 separate reads. However, at this coverage, nearly 50% of sequence variants identified are false positives due to mapping errors, polymerase errors, and the low frequency of somatic variants within the sample. Therefore, secondary validation of variants remains a necessary part of all sequencing projects. The detection of small insertions and deletions (indels) using current algorithms is problematic. In addition, mapping sequence reads to the approximately 50% of the human genome that contains repetitive sequences is imperfect, raising the possibility that mutations are being missed. The incident AML case highlighted these technical issues11 ; a single base-pair deletion in DNMT3A was missed during short-read analysis (30- to 40-bp reads, which are difficult to align to the reference genome if they contain an indel), but was later identified when the genome was resequenced with paired-end reads (pairs of 50- to 75-bp reads, which align more robustly). These problems will be improved as bioinformatics can draw on additional sequenced human genomes and as decreased costs permit deeper sequencing, but efficiently mapping sequences to repetitive regions of the genome will likely require technical improvements that permit longer sequence read lengths.
There are inherent limitations of whole-genome sequencing that need to be considered when analyzing cancers. First, each AML genome carries approximately 500-1000 somatic mutations.12 It is likely that the majority of these mutations do not contribute to leukemic transformation. Identifying the “driver'” mutations in each AML genome is currently challenging. Second, whole-genome sequencing does not provide information about epigenetic modifications and alterations in gene expression. Therefore, to fully characterize acquired genomic changes in an AML patient, assays to measure RNA expression (eg, transcriptome sequencing) and epigenetic changes (eg, bisulfate sequencing to define the methylome) in combination with whole-genome sequencing are required.
Genome sequencing remains expensive, and the requisite infrastructure, expertise, and time to complete sequence analysis are significant barriers to the routine use of whole-genome sequencing of AML in the clinical setting. However, as the number of sequenced AML genomes increases, the technical and bioinformatic infrastructure required for clinical analysis will become more accessible. Current cost estimates at the Washington University Genome Institute for whole-genome sequencing of a paired normal/leukemia sample is $40 000. This is a fully loaded cost that includes sequencing, bioinformatic analysis, and validation. The current time to sequence and analyze a paired tumor and normal genome is 6 weeks. The cost of whole-genome sequencing needs to be considered in the context of the increasingly complex diagnostic evaluation for AML. A common molecular evaluation consists of cytogenetics; FISH for t(15;17), t(8;21), inv(16), and t(9;22); and molecular testing for FLT3, KIT, and NPM1 mutations. The list price of this “standard of care” molecular evaluation at Quest Diagnostics (a national laboratory) is currently $4575 and does not include the cost of molecular testing for other recurring mutations (eg, NRAS, IDH1, IDH2, TET2, DNMT3A, RUNX1, ASXL1, and CEBPA) that are likely to become routine. As the cost of sequencing continues to decrease and the number of molecular assays continues to increase, we will rapidly reach the inflection point where whole-genome sequencing will be the most cost-effective diagnostic tool for AML patients.
Finally, there are important practical and ethical considerations concerning the clinical application of whole-genome sequencing. Based on the cancer genomes analyzed to date, it is clear that a tremendous number of genetic variants will be identified in each AML genome, of which the majority will be inherited variants. Some of these will be in genes known to be involved in cancer susceptibility, and some will be associated with unsuspected genetic diseases (eg, hereditary hemochromatosis). Best practices to communicate this information to patients and their families are being considered and developed by several groups of investigators.
Potential clinical applications (current and future)
Whole-genome sequencing provides a method to identify all genetic changes in a cancer genome, including single-nucleotide mutations, deletions, amplifications, chromosomal translocations, and uniparenteral disomy, at a single-base resolution. There are several ways that this unprecedented ability to characterize cancer genomes may affect the clinical management of patients with AML.
Classification of AML
Whole-genome sequencing may lead to a better classification of AML. There is evidence that a molecular classification of AML based on karyotype and specific gene mutations is superior to a morphology-based classification. As additional mutations are discovered in AML, it is likely that the molecular classification of AML will be further defined. Having access to all of the genetic changes in an AML genome through whole-genome sequencing would facilitate this process.
Optimizing therapy of AML
A detailed knowledge of mutations present in the AML genome may help to optimize the therapeutic plan. For example, mutations in tyrosine kinase genes may suggest the use of specific small-molecule inhibitors. In addition, disruption of specific pathways in the AML genome may be exploited therapeutically. For example, mutations in BRCA1 or BRCA2, by inhibiting DNA repair through homologous recombination, may render cancer cells more sensitive to inhibitors of protein poly(ADP)-ribose polymerase.28 Finally, the discovery of new mutations in AML may lead to the development of novel targeted therapeutics.
Pharmacogenomics
Pharmacogenomics is the study of how an individual's genes influence his or her response to drugs. Genetic variants can influence the metabolism, toxicity, and therapeutic efficacy of a drug. By sequencing the “normal” and leukemia genomes in patients with AML, genetic variants known to effect the efficacy and toxicity of chemotherapeutic agents will be identified and this information may affect the choice and dose of chemotherapy used. For example, germline polymorphisms in the UDP glucuronosyltransferase 1 family (UGT1A1) increase the toxic effects of irinotecan,29 and germline polymorphisms in thiopurine S-methyltransferase (TMPT) increase sensitivity to mercaptopurine through decreased metabolism.30 As more genomes are sequenced, the list of genetic variants influencing response to chemotherapy likely will expand.
Cancer susceptibility
As discussed above, whole-genome sequencing of the “normal” genome in patients with AML may identify genetic variants contributing to cancer susceptibility. Not only will this information be important for genetic counseling, it may affect the patient's treatment and prognosis. Whole-genome sequencing will provide genotyping data for the more than 100 genes already implicated in cancer susceptibility. Causal associations between germline variants and AML have been firmly established for only a few genes, such as TP5331 and RUNX1.32 However, it is likely that the list of genetic variants contributing to AML susceptibility will increase in the near future as additional genomes are sequenced.
Conclusions
The Human Genome Project and advances in sequencing technologies have revolutionized our ability to characterize cancers at a molecular level. Using whole-genome sequencing, it is now possible to interrogate cancers genome wide at a single-base-pair resolution. Application of whole-genome sequencing to AML has already yielded important discoveries, including the identification of common gene mutations in AML (eg, IDH1 and DNTM3A). Ongoing whole-genome sequencing of additional AML samples likely will identify the majority of relevant mutations in AML. In the near term, sequencing of a panel of AML-related medically actionable mutations could be performed economically using next-generation sequencing and custom-capture arrays. However, with continued improvements in sequencing, it is likely that whole-genome sequencing will become a routine part of the diagnostic workup of patients with AML and other cancers.
Disclosures
Conflict-of-interest disclosure: J.S.W. has received research funding from Eisai Pharmaceuticals. D.C.L. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Daniel C. Link, MD, Division of Oncology, Department of Medicine, 660 S Euclid Ave, Campus Box 8007, St Louis, MO 63110; Phone: (314) 362-8771; Fax: (314) 362-9333; e-mail: dlink@dom.wustl.edu.
