
Abstract
Peripheral T-cell lymphomas (PTCLs) encompass a group of rare and usually clinically aggressive diseases. The classification and diagnosis of these diseases are compounded by their marked pathological heterogeneity and complex clinical features. With the exception of ALK-positive anaplastic large cell lymphoma (ALCL), which is defined on the basis of ALK rearrangements, genetic features play little role in the definition of other disease entities. In recent years, hitherto unrecognized chromosomal translocations have been reported in small subsets of PTCLs, and genome-wide array-based profiling investigations have provided novel insights into their molecular characteristics. This article summarizes the current knowledge on the best-characterized genetic and molecular alterations underlying the pathogenesis of PTCLs, with a focus on recent discoveries, their relevance to disease classification, and their management implications from a diagnostical and therapeutical perspective.
Introduction
Peripheral T-cell lymphomas (PTCLs), a collective term to designate malignancies derived from mature (post-thymic) T cells and natural killer (NK) cells, encompass a heterogeneous group of rare diseases, altogether accounting for < 15% of all cases of non-Hodgkin lymphoma worldwide.1
In contrast to the major progress made in the management of B-cell lymphomas over the past years, advances in the field of T-cell lymphomas have been much slower, and dealing with this category of diseases currently remains a challenge from both the therapeutical and pathobiological perspectives. With few exceptions, most entities are clinically aggressive diseases with overall poor responses to classical treatments and a dismal prognosis.1 The identification of PTCL is often difficult because many cases comprise a reactive cellular infiltrate that may mask the neoplastic cell population. Moreover, demonstration of T-cell clonality is not feasible by routine immunophenotyping and usually requires molecular testing. With respect to the multiparametric approach to lymphoma diagnosis as developed in the World Health Organization (WHO) classification, which is based on a combination of morphologic, immunophenotypic, genetic, and clinical features, and correlation to a normal cellular counterpart,2 its application to NK-T cell–derived neoplasms is hampered by several difficulties. There is significant morphologic and immunophenotypic overlap across different entities while conversely the clinical features and anatomic location of the disease are critical in defining disease entities, which are grouped according to their presentation as disseminated, predominantly extranodal or cutaneous, or predominantly nodal diseases (Table 1). At variance with other categories of hematologic neoplasms, in which cytogenetic analyses have been instrumental in identifying recurrent chromosomal translocations, thereby elucidating their pathogenesis and providing information relevant to their diagnosis and prognosis, only few recurrent genetic alterations have been identified in PTCLs. In recent years, however, novel insights have been gained from genome-wide array-based molecular genetic technologies.
Mature T- and NK-cell neoplasms in the 2008 WHO classification of lymphoid tumors

*Provisional entity.
(Adapted from Swerdlow et al.2 )
This article summarizes the current knowledge on the best-characterized genetic and molecular alterations underlying the pathogenesis of PTCLs, with a focus on recent discoveries. Their relevance to the classification and diagnosis of these heterogeneous tumors, and their implications for the development of novel therapeutic approaches and clinical management, are also discussed.
Recurrent chromosomal translocations
ALK translocations
Translocations involving the anaplastic lymphoma kinase (ALK) gene on chromosome 2 represent the most well-established recurrent translocation among PTCLs. The most common ALK translocation is t(2;5)(p23;q35), which was first described by several groups in the late 1980s and fuses the ALK to the nucleophosmin gene (NPM1) on chromosome 5.3,4 Since then, several other translocations in which the ALK gene is fused to other partner genes have been reported (Table 2), and it is likely that additional infrequent translocations will be recognized in the future. The different fusion proteins generated by the translocations share a common structure comprising the 5′-end partner fused to the ALK tyrosine kinase domain at the 3′ end. These chimeric fusion proteins contain dimerization domains provided by the partner gene, which induce constitutive activation of tyrosine kinase ALK.5
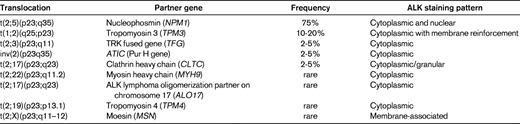
Within the spectrum of PTCLs, a large number of primary systemic anaplastic large cell lymphomas (ALCLs; 50%-85%) carry ALK translocations and overexpress the ALK protein. ALK-positive ALCL is currently the sole PTCL entity in the WHO classification that is defined on a genetic basis, by ALK gene rearrangement resulting in ALK overexpression.2 This well-defined entity comprises a morphologic spectrum, including a common pattern characterized by anaplastic large cell morphology with so-called “hallmark cells”, strong CD30 expression, a tendency for a sinusoidal and cohesive growth pattern, and several variant patterns (tumors with predominantly small cell morphology, a lymphohistiocytic variant comprising numerous reactive histiocytes, and the giant cell and sarcomatoid variants). Consequently, demonstration of deregulated ALK is the key to the diagnosis and identification of ALK-positive ALCL. Polyclonal and monoclonal antibodies directed at the intracellular portion of ALK react with ALK fusion proteins and the full-length ALK protein, which is normally not expressed in the lymphoid tissues. Therefore, immunohistochemistry is routinely used to detect lymphoid cells expressing ALK fusion proteins. Interestingly, the subcellular distribution of the staining depends on the type of translocation (Table 2). The most common t(2;5) translocation is the only one to produce ALK staining in the nucleus and cytoplasm, whereas in variant translocations usually only cytoplasmic staining is observed. Chromosomal translocations involving breaks at 2p23 may be detected on standard karyotypes, a procedure that is inconsistently applied in routine diagnosis, or by FISH with break-apart probes. RT-PCR assays are in general designed for the detection of NPM1-ALK transcripts and not for variant translocations. Therefore, in general, immunohistochemistry has supplanted molecular tests for the diagnosis of ALCL because of its sensitivity and specificity. In the case of lymphomas composed of large cells with a T-cell or null immunophenotype and CD30 expression, ALK testing is mandatory to segregate the subset of ALK-positive ALCLs from other ALK-negative CD30+ T-cell lymphomas, a distinction that is clinically much relevant because ALK-positive ALCL (irrespective of the type of translocation) carries a much better prognosis than both ALK-negative ALCLs and PTCL, not otherwise specified (PTCL, NOS).6 In the case of ALK-positive ALCLs with variant morphology, in which CD30 expression may be restricted to a subset of the neoplastic cells, ALK testing is also mandatory in establishing the diagnosis and is critical in identifying the neoplastic nature of the lymphoproliferation in some of the cases, which may be overlooked as reactive by morphology alone. There is no correlation between the morphologic patterns and the type of translocation.
ALK translocations and overexpression are not specific for ALCL, because tumors other than ALCL can express ALK either as fusion proteins or as full-length proteins (Table 3).7 Native ALK protein is expressed in a subset of rhabdomyosarcomas and in most neuroblastomas, which may harbor ALK point mutations or amplifications.8 ALK-positive large B-cell lymphomas represent a rare subset of diffuse large B-cell lymphomas with plasmablastic features, lack of CD30 expression, poor response to chemotherapy, and frequently fatal outcome. Most cases express ALK fused to clathrin as a consequence of a t(2;17) translocation, but other translocations overlapping with those encountered in ALK-positive ALCL occur as well.9–11 ALK rearrangements also occur in ∼1/3 of inflammatory myofibroblastic tumors. In addition, a translocation fusing ALK to echinoderm microtubule-associated protein like 4 (EML4) has been described more recently in a small subset of lung adenocarcinomas, implying that this fusion protein functions as an oncogenic driver and ALK inhibitor (crizotinib) as a specific targeted therapy in these carcinomas.12
ALK-positive neoplasms
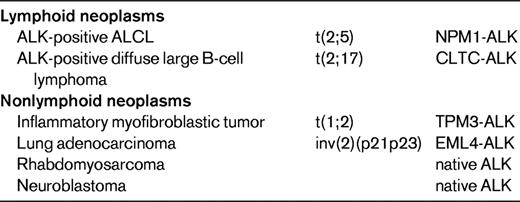
For each type of neoplasm, the most common form of ALK protein expressed is listed.
CLTC indicates clathrin heavy chain; EML4, echinoderm microtubule associated protein like 4; NPM1, nucleophosmin; and TPM3, tropomyosin 3.
Numerous studies have proven that NPM1-ALK is oncogenic.5,13 In vitro, constitutively activated ALK chimeras induce cellular transformation, enhance cell proliferation and survival, and lead to cytoskeletal rearrangements and changes in cell shape. Oncogenic ALK transformation is mediated by interaction with downstream molecules that engage intracellular signaling pathways, the most relevant and better characterized being the ERK, the JAK3-STAT3, and the PI3K-Akt pathways. In ALCL cell lines, STAT3 expression has been demonstrated as the master mediator of ALK-induced gene regulation.14 Notably, the loss of expression of T-cell-specific molecules including TCR-related signaling molecules, results from STAT3-mediated gene-transcription regulation and/or epigenetic silencing.15 Whether the loss of TCR signaling molecules could give a survival advantage to ALCL cells remains an open question. CAAT/enhancer binding protein beta (CEBPB) is another critical ALK-regulated target gene necessary to induce cell transformation and sustain the growth and survival of ALK-positive ALCL cells in vitro.16–18 NPM1-ALK transgenic mice that spontaneously develop precursor T-cell lymphomas have been established, and in this in vivo model, lymphomagenesis is dependent on STAT3. Interestingly, enforced expression of NPM1-ALK in transgenic models also leads to B-cell transformation and induces B-cell lymphomas or plasma cell neoplasms. Moreover, the t(2;5) translocation is detected at low rates in healthy individuals, and has been recorded in cord blood,19 a finding that may be correlated with the main peak of occurrence of ALK-positive ALCL in childhood, but it also implies that ALK rearrangements are not sufficient for lymphoma development, and indeed secondary genetic alterations are common.20
T(5;9)(q33;q22) translocation
The t(5;9)(q33;q22) translocation fusing the IL-2-inducible T-cell kinase (ITK) gene on chromosome 5 with the spleen tyrosine kinase (SYK) gene on chromosome 9 was described in 2006 by Streubel et al.21 This genetic abnormality represents the first recurrent chromosomal translocation in PTCL, NOS, which is found to occur in a small minority of the cases, predominantly in association with tumors of peculiar morphology with so-called follicular features.21 Follicular PTCL (F-PTCL), named after a pattern of growth intimately related to follicular structures, is a rare variant of PTCL that comprises cases with a truly follicular pattern that mimic follicular lymphoma22 and T-cell lymphomas with a perifollicular growth pattern23 or involving expanded mantle zones.24 F-PTCL is currently listed as a variant of PTCL, NOS in the WHO classification;however, the neoplastic cells in F-PTCL usually display an extensive follicular helper T-cell immunophenotype and few cases have been reported in association with angioimmunoblastic T-cell lymphoma (AITL), suggesting a closer relationship to AITL.25
The t(5;9) translocation, which is detected by FISH in ∼20% of F-PTCL cases, is otherwise rarely incriminated in nonfollicular PTCLs and has not been detected in AITL, overall supporting the distinction of F-PTCL as a PTCL variant.21,25 In daily practice, however, demonstration of the translocation, whereas ascertaining the diagnosis of malignancy and representing a further element to support the diagnosis in cases of F-PTCL, is of limited utility and the biological implications in a given tumor remain unclear.
The ITK-SYK fusion gene compromises the 5′-coding sequence of ITK joined to the 3′-coding sequence of SYK and encodes a kinase-to-kinase chimeric protein in which the N-terminal pleckstrin homology and TEC homology domains of ITK are fused to much of the interdomain B and the complete C-terminal kinase domain of SYK.21 ITK-SYK is a catalytically active tyrosine kinase with transforming properties in vitro, as demonstrated in NIH3T3 cells.26 The membrane localization of the fusion protein is critical to its transforming properties, and is dependent upon PI3K signaling.26,27
In vivo, the oncogenic properties of the overexpressed fusion gene have been shown in a transgenic mouse model expressing ITK-SYK fusion under the control of a T cell–specific promotor, and in another mouse model transplanted with BM cells transduced with a vector expressing ITK-SYK.28,29 In both models, overexpression of the chimera resulted in the formation of highly malignant PTCLs, with a phenotype resembling that described in human patients. In T cells from transgenic mice, ITK-SYK fusion translocates to lipid rafts and triggers antigen-independent phosphorylation of TCR-associated signaling proteins, indicating that enforced antigen receptor signaling can function as a powerful oncogenic driver.28
In contrast to the rare prevalence of the SYK-ITK translocation, overexpression and activation of SYK, which is normally present in immature T cells and is down-regulated at the mature stage, have been recently reported as a feature common to most PTCLs. This may represent a novel therapeutic target, because inhibition of SYK induces apoptosis and blocks proliferation in T-cell non-Hodgkin lymphoma cell lines.30,31
Translocations involving TCR gene loci
There are 3 genetic loci for TCR genes: alphadelta (TRA@/TRD@ at 14q11), beta (TRB@ at 7q34), and gamma (TRG@ at 7p14). Because the delta locus is situated entirely within the alpha locus, they are considered together. Translocations involving the TCR genes in T-cell lymphomas may induce the transcription of an oncogene on the partner chromosome, a mechanism that is commonly operating in precursor T-cell lymphoblastic neoplasms. Among mature T-cell tumors, such a mechanism is typically incriminated in T-cell prolymphocytic leukemia (T-PLL), but has been otherwise rarely found in other mature T-cell neoplasms.
The majority (>90%) of T-PLL cases are characterized by translocations or inversions with the TRA@ gene. The genes involved in the rearrangement are (by decreasing frequency) the TCL1 and TCL1b genes at 14q32—inv(14)(q11;q32.1) or t(14;14)(q11;q32.1)—or the MTCP1 gene at Xq28—t(X;14)(q28;q11)—which are overexpressed as a consequence of juxtaposition to the TRA@ locus and encode proteins with similarities of structure.32 TCL1 is a cytoplasmic protein with the property of binding to AKT1, enhancing its activity and promoting its transport to the nucleus; it is normally expressed in early T-cell progenitors and in mature B cells, but is absent in mature T lymphocytes. Overexpression of TCL1 in T-PLL, detected by strong nuclear positivity on immunostaining, confers resistance to activation-induced cell death and growth arrest in T-PLL cells and derived cell lines. The oncogenic properties of the rearrangement are illustrated by the fact that transgenic mice overexpressing either activated TCL1 or MTCP1 gene in T cells develop mature T-cell leukemias. TCL1 gene rearrangements are specific for T-PLL and are not observed in other categories of T-cell neoplasms.33
Chromosomal breaks involving the TCR gene loci (mostly the TRA@ locus) have been reported in rare cases of PTCL, NOS, and their overall prevalence in several FISH-based screening studies is extremely low. The highest frequency was reported by Nelson et al, who found rearrangements at the TRA@/TRD@ locus in 5 of 58 PTCL, NOS cases, perhaps an overestimate because only cases with an abnormal karyotype were analyzed in that study.34
These TCR translocations remain poorly understood because the identity of the translocation partner(s) has not been identified in most cases.33–36 PVRL2 (poliovirus receptor-related 2) has been cloned as the partner gene of the TRA@ in the t(14;19)(q11;q13) translocation and appears to be associated with overexpression of both PVRL2 and BCL3 mRNAs.37,38
Recently, involvement of the multiple myeloma oncogene-(MUM1)/IFN-regulatory factor-4 (IRF4) oncogene locus in chromosomal translocations involving the TRA@ gene, t(6;14)(p25;q11.2), was identified in 2 cases of PTCL, NOS with a cytotoxic phenotype and a peculiar clinical presentation characterized by involvement of the bone marrow and skin without significant lymphadenopathy or hepatosplenomegaly. The marked similarity between the 2 cases with IRF4/TRA@ translocation suggests that they may represent a distinct clinicopathologic entity.39
Translocations involving the MUM1/IRF4 oncogene locus (6p25.3)
After the discovery of translocations leading to IRF4/TRA@ fusions in occasional PTCL, NOS cases, FISH screening of a large panel of PTCL entities for IRF4 rearrangements identified non-TCR-related IRF4 translocations in additional cases, mostly ALK-negative ALCLs, of the primary cutaneous or primary systemic types, with an estimated overall prevalence of 28% and 18%, respectively.39–42
Among primary cutaneous T-cell lymphoproliferative disorders, FISH positivity for an IRF4 translocation is highly specific for primary cutaneous ALCL (99% specificity), because only occasional cases of lymphomatoid papulosis (1 of 39 cases) or transformed mycosis fungoides (2 of 24 cases) have an IRF4 rearrangement, which was otherwise not found in other T-cell lymphoproliferative disorders involving the skin (although it could theoretically be positive in cases of cutaneous dissemination of systemic ALK-negative ALCL).40,41 Conversely, other IRF4 FISH abnormalities, mainly extra copies of the IRF4 locus mutually exclusive with translocations, also occur and are more widely distributed over the T-cell lymphoproliferation subtypes.41 Because primary cutaneous ALCL may be difficult to distinguish from other CD30-positive lymphoproliferations in the skin, and given the management consequences of a correct diagnosis, FISH testing for IRF4 rearrangements is of clinical utility to assist in the differential diagnosis of primary cutaneous T-cell lymphoproliferations in conjunction with morphological, immunophenotypical, and clinical features.41
Interestingly, the rearrangements of the IRF4 locus on 6p25.3 correspond to break points involving the IRF4 gene in only ∼1/3 of the cases, and the levels of IRF4 mRNA and the detection of IRF4 protein by immunohistochemistry are similar in translocated and nontranslocated ALK-negative ALCLs.40–42 More commonly, the break points at 6p25.3 involve the DUSP22 phosphatase gene located immediately telomeric to IRF4. Using a combination of mate-pair DNA library construction, massively parallel (“next-generation”) sequencing, and an original bioinformatics algorithm, Feldman et al recently identified the FRA7H fragile site on 7q32.3 as a recurrent gene partner of DUSP22 on 6p25.3 in a balanced t(6;7) translocation. In all, the t(6;7)(p25.3;q32.3) translocation accounts for ∼1/3 of IRF4 locus rearrangements in ALK-negative ALCLs.42 The translocation occurs in both systemic and cutaneous ALK-negative ALCLs and therefore may contribute to some of the common features between these 2 entities. Among PTCLs, the 7q32.3 break point is specifically involved in the t(6;7)(p25.3;q32.3) translocation, usually with DUSP22 as a gene partner, but IRF4-FRA7H rearrangements appear to also occur, albeit uncommonly. Conversely, FRA7H is not the only locus involved in translocations with break points at 6p25.3.
DUSP22 is a dual-specificity phosphatase that inhibits T-cell antigen receptor signaling in T cells by inactivating ERK2 MAPK. As a consequence of the t(6;7) translocation, DUSP22 is down-regulated, suggesting its role as a tumor suppressor gene, whereas there is up-regulation of MIR29 miRNA on 7q32.3, which may have an oncogenic function, as has been proposed in other cancer models.
It remains unclear whether rearrangements or other abnormalities of the IRF4 locus bear a prognostic significance.
Other chromosomal abnormalities
Isochromosome 7q
Isochromosome 7q, or i(7)(q10), is a recurrent chromosomal aberration in hepatosplenic T-cell lymphoma that is observed in the majority of the cases, both those with a gammadelta phenotype and those with an alphabeta phenotype.43,44 Isochromosome 7q results in the deletion of the short arm of chromosome 7, which may lead to the loss of tumor suppressor genes located on 7p, as well as loss of TRB@ gene at 7p15. Duplication of the long arm most likely causes overexpression of oncogenes located on 7q, as well as the TRG@ gene at 7q35.44 In rare instances, extra copies of 7q are caused by ring chromosome 7.45 Isochromosome 7q is usually thought to be the primary abnormality of this disease with a tendency to multiply the i(7)(q10) during disease progression; it may be accompanied by trisomy 8 and loss of a sex chromosome, which also seem to be associated with progression of the disease.44,46,47 Isochromosome 7q is detected by standard cytogenetics and is also amenable to investigation by FISH using a 2-color probe assay to 7p and 7q. Isochromosome 7q is not specific for hepatosplenic T-cell lymphoma, being one of the most common isochromosomes in malignant disorders (acute myeloid and lymphoblastic leukemias, myelodysplastic syndromes, and Wilms tumor). In the spectrum of NK/T-cell malignancies, isochromosome 7q has also been found on occasion in cases of NK/T-cell lymphomas and ALK-negative ALCLs.33
Deletion 6q
Deletion at 6q21-25 is the most recurrent chromosomal aberration in extranodal, nasal-type NK/T cell lymphoma (NKTCL), a distinct entity more frequent in Asian and South-American populations characterized by a proliferation of EBV-infected NK neoplastic cells, usually in the nasopharynx and a dismal prognosis. The 6q deletion is observed in the majority of the cases, in association with less frequent gains at chromosomes 1p, 6p, 11q, 12q, 17q, 20q, and Xp and losses at 11q, 13q, and 17p, indicating a complex karyotype.48–52 Losses at 6q21-q27 appeared more frequently in NKTCL than in aggressive NK-cell leukemia, a rare disease regarded as the systemic variant of NKTCL.53 Two recent studies comparing high-resolution array-based comparative genomic hybridization and gene-expression profiling on NK cell lines and NKTCL tumor samples emphasized recurrent losses of 6q21 and 17p11.2-p13.3 and gain of 1q21-q44 in approximately one-half of NKTCL patients.54,55 The region of del6q21 contains 4 candidate tumor suppressor genes, PRDM1, ATG5, AIM1, and HACE1. These 4 genes show decreased expression confirmed at the RNA level in NKTCL cell lines and/or NKTCL tumors, which has been attributed to mutations and methylations in PRDM1, ATG5, and AIM1 observed in NKTCL cell lines.54,55 Therefore, loss-of-function due to mutations and transcriptional inhibition by DNA methylation suggest a tumor-suppressor role for these genes in NKTCL.54
Chromosome 9 and chromosome 1 abnormalities
Enteropathy-associated lymphoma (ETL), a rare T-cell lymphoma entity that derives from intestinal intraepithelial lymphocytes and appears to be a complication of gluten-sensitive enteropathies, is characterized by frequent complex gains of 9q31.3-qter (or by an almost mutually exclusive 2.5-megabase loss of 16q12.1 in 23% of cases). The 9q alteration is seen in both the common type of ETL (type 1) and in a rare monomorphic variant (type 2) in approximately 70% of cases.56,57 This region contains 2 candidate genes, ABL1 and especially NOTCH1, which are preferentially amplified in ETL.58 Aberrations at 9q33-34 appear, however, not completely specific, being also found in around 20% of PTCL, NOS.59
A recurrent structural partial trisomy 1q22-q44 is also found in refractory sprue, which is regarded as a first step toward malignant transformation and has been proposed as a frequent early event.60
Deletions/loss of heterozygosity at chromosome 9p21, which harbors the tumor-suppressor genes CDKN2A and CDKN2B, are frequent in type 1 ETL, especially in tumors with large cell morphology. Loss of heterozygosity most frequently affects the region spanning the p14/p15/p16 gene locus and is correlated with the loss of p16 expression in these cases, suggesting that gene loss at this locus may play a role in the development of ETL.61
Other chromosomal aberrations
By standard cytogenetics, abnormal karyotypes are variably found in PTCLs.62 In PTCL, NOS, complex karyotypes are common, especially in cases with large cell morphology. In AITL, recurrent karyotypic aberrations include trisomies of chromosomes 3, 5, and 21, gain of X, and loss of 6q.34
In PTCL, NOS,34,59,63 recurrent chromosomal gains have been observed by comparative genomic hybridization in chromosomes 7q (targeting cyclin-dependent kinase 6),64 8q (involving the MYC locus),63 17q, and 22q, and recurrent losses in several chromosomes. In one study, deletions in chromosomes 5q, 10q, and 12q were associated with a better prognosis.59
Molecular signatures
Over the past years, several groups have reported on the molecular characterization of PTCLs by gene-expression profiling (GEP) methods applied to primary tumors representative of the various PTCL entities and cell lines, with comparison with reactive tissues and sorted normal NK/T–cell populations.14,18,54,55,62,65,66,70 In general, molecular profiles have shown variable concordance to the pathological classification, with some overlap between entities, especially between PTCL, NOS and other nodal PTCLs, which may reflect the influence of nonneoplastic elements or the existence of common tumor-associated pathways. In addition, GEP studies have provided significant novel insights into the pathogenesis and molecular mechanisms underlying PTCL development, which may be relevant to the development of novel therapeutic approaches.
Cellular origin of PTCL entities
Application of the WHO classification principles to T-cell neoplasms has been impaired in part by the imprecise characterization of their cellular derivation. A major recent advance in that field was the identification of the normal cellular counterpart of AITL, one of the most common PTCL entities. GEP analyses have been critical in establishing molecular similarities between the AITL neoplastic component and a peculiar subset of CD4+ T cells normally located in germinal centers and exerting helper functions to follicular B cells, the so-called follicular helper T cells (TFH cells).65,66 The characterization of AITL as a TFH-derived neoplasm not only accounts for pathobiological features of this disease entity,67 but also has important implications for AITL diagnosis and classification. Indeed, several markers of normal TFH cells, including the CXCL13 chemokine and several cell-surface markers (eg, PD1, ICOS, and CD200) have been validated as novel diagnostic biomarkers of AITL that are useful in assisting in the differential diagnosis with other PTCL entities.68 Moreover, traces of the TFH signature have been identified among cases originally classified as PTCL, NOS, suggesting that the AITL spectrum may be wider than suspected.65,68,69
At the molecular level, PTCL, NOS cells appear to be most closely related to either CD4 or CD8+ activated T cells, and are characterized by deregulation of genes related to proliferation, apoptosis, cell adhesion, and matrix remodeling, which is similar to many other malignancies.70 The heterogeneity of PTCL, NOS has also been highlighted at the molecular level, with a molecular group comprising cases rich in histiocytes (including the so-called lymphoepithelioid variant) and a small molecular subgroup with cytotoxic features and a poor prognosis, delineated in the study by Iqbal et al.71
Molecular pathways
GEP has identified distinct gene clusters reflecting biological features related to the neoplastic cells and/or the reactive cell populations that may be relevant to the delineation of molecular subgroups. Classification according to differential expression of NF-κB pathway genes may be clinically useful, because overexpression of this pathway was found to be correlated with a better outcome.72 Conversely, overexpression of a proliferation signature was correlated with an adverse prognosis, and appears to be inversely related to signature clusters related to inflammatory response and reflective of an abundant reactive background in the tumor.73,74
Conclusions
From this summary on the current state on genetic and molecular features of PTCLs, it is apparent that a significant number of new findings have accumulated over the past years. The recent discovery of novel recurrent chromosomal translocations in association with PTCL subsets represents a major advance in the understanding of the pathogenic mechanisms underlying these disorders, and it may be foreseen that others remain to be discovered, especially in the context of the current developments in “deep sequencing” methods and in bioinformatical algorithms. Altogether, recent genetic discoveries and molecular profiling studies have helped to refine the criteria for the classification and diagnosis of several PTCL disease entities, and have already led to the development of novel immunophenotyping diagnostic tools as surrogates for molecular signatures (Table 4 and Figure 1). The indications for genetic and/or molecular testing in routine diagnosis presently remain limited, but are expected to expand in light of the development of targeted therapies.


Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Laurence de Leval, MD, PhD, Service de Pathologie Clinique, Institut de Pathologie, CHUV, Université de Lausanne, 25 rue du Bugnon, CH-1011 Lausanne, Switzerland; Phone: 41-21-3147194; Fax: 41-21-3147205; e-mail: Laurence.deLeval@chuv.ch.
