
Abstract
Consideration of primary prophylaxis to prevent complications of sickle cell disease (SCD) requires a clear understanding of the earliest manifestations of SCD-related organ injury, a reliable clinical or laboratory tool to detect organ dysfunction, and evidence that an intervention instituted in the presymptomatic state can mitigate disease progression. This review examines the pathophysiology of SCD in organs that may be potential targets for intervention, our current capacity to evaluate early SCD manifestations, results of clinical trials, and opportunities for future interventions.
Introduction
Sickle cell disease (SCD) encompasses inherited anemias due to beta globin mutations that result in the formation of sickled red cells and increased red cell turnover. The complications of SCD are both acute and chronic, and this combination results in significant morbidity, high healthcare utilization over the lifespan, and increased premature death.1 Whereas many avenues have been explored to manage complications of SCD as they arise, renewed attention has been focused on primary prevention before signs of organ dysfunction have appeared, particularly in young children. This review highlights areas of established benefit of primary prophylaxis and emerging data on interventions that may ultimately change the natural history of this condition for patients. Acute and recurrent sickling events ultimately result in organ damage and impaired function. Understanding the pathophysiology and timing of this organ-specific progression, as well as early clinical or laboratory indicators of injury, is necessary to determine: (1) which processes might be amenable to primary prevention, (2) when interventions should take place, and (3) what medications or procedures might be effective. Secondary prophylaxis will only be reviewed as it relates to making a case for intervention in the presymptomatic state. Table 1 provides a summary of the potential targets for primary prophylaxis in SCD.
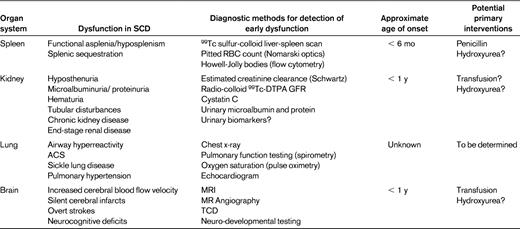
Organ-specific targets for primary prevention in SCD: pathophysiology
Spleen
Splenic function is key for maintaining normal immune homeostasis, including opsonization of pathogens such as invasive encapsulated organisms, phagocytic filtration of senescent or damaged cells, and maintenance of lymphocyte subpopulations necessary for both innate and adaptive immune responses.2 Vesicles that accumulate on the RBC surface with age are normally removed by the intact spleen, but these pocked or pitted RBCs remain visible in the circulation in individuals with hyposplenism or asplenia. The developmental patterns of hyposplenism or functional asplenia in SCD were described by Pearson and the other investigators of the Cooperative Study of Sickle Cell Disease (CSSCD) using radionuclide spleen scans and pocked RBC counts.3 A cohort of infants were first evaluated at 8-13 months of age and nearly 69% had pocked RBC counts < 3.5% and simultaneous splenic radio-colloid uptake classified as normal. Those with normal uptake tended to be younger, with higher fetal hemoglobin levels and less anemia. Pocked RBC data on the overall CSSCD population demonstrated that the mean pocked RBC percentages increased most rapidly in the first 6 months of life for individuals with homozygous SCD, somewhat more gradually with the other sickle hemoglobinopathies, and that this timing correlated well with epidemiologic data on the incidence rates of severe bacterial infections. Quantitative detection of Howell-Jolly bodies by flow cytometry has also been used to assess splenic dysfunction. Once autoinfarction of the spleen occurred, it was presumed that splenic function was irreversibly lost.
More recently, the baseline data on splenic function from a larger infant SCD cohort in the Pediatric Hydroxyurea Phase III Clinical Trial (BABY HUG) found a similar, strong correlation of pocked or pitted RBC counts with liver-spleen scans; however, a much smaller percentage of infants had normal uptake (23%) compared with CCSCD.4 Apparent absent or decreased splenic function was significantly correlated with older age, lower total hemoglobin, lower fetal hemoglobin, and higher WBC. Measurements of Howell-Jolly bodies by flow cytometry were also correlated with the pitted RBC and scintigraphic assessments of splenic function at baseline. By age 12 months, 84% of the infants with SCD in this cohort had decreased or absent splenic uptake.
Kidney
Glomerular hyperfiltration is common in children with SCD. Hyposthenuria and asymptomatic microalbuminuria may be among the earliest signs of renal impairment,5 and limited data suggest that detection of microalbuminuria, defined as urine albumin of 30-300 mg/g of creatinine, predicts the course toward further glomerular injury.6,7 Increased renal plasma flow and intraglomerular hypertension are associated with tubular defects that result in decreased urinary concentrating ability.8 Persistent proteinuria occurs in 6%-12% of pediatric patients with SCD9 as early as 7 years of age, with increasing prevalence into early adulthood.10 Over time, recurrent intrarenal sickling results in sickle nephropathy with features most consistent with focal segmental glomerulosclerosis due to both hyperfiltration and glomerular hypertrophy. SCD-related chronic kidney disease may lead to end-stage renal disease requiring dialysis or transplantation in up to 30% of adults. Well-defined screening and diagnostic tools are needed.
Accurate assessment of the glomerular filtration rate (GFR) is key to identifying opportunities for prevention or early intervention. Estimation of GFR based on the calculated creatinine clearance using the Schwartz or modified cystatin C formula typically overestimates filtration capacity in SCD. Quantitative methods such as inulin clearance or technetium-labeled diethylene-triaminepentaacetic acid (99Tc-DTPA) may be more accurate but may also have limitations in SCD, particularly at the upper end of the range.
The baseline observations of the BABY HUG study included assessment of renal function.11 At study entry, 77.2% of infants at a mean age of 13 months in BABY HUG were able to concentrate urine with controlled fluid deprivation.12 In addition, these young children had scintigraphic GFR measures well above normal for age, suggesting that the renal impairment that leads to glomerular hyperfiltration occurs very early in life. Calculated GFR estimates using the Schwartz equation in general were higher, more variable, and only weakly correlated with renal clearance by 99Tc-DTPA. Quantitated GFR by DTPA in this cohort was correlated with age, height, weight, kidney volume, and decreased serum creatinine, but not with total or fetal hemoglobin. Although a direct progressive relationship of early hyperfiltration to later renal manifestations of SCD has not been fully elucidated, the early onset of impairment is nonetheless concerning and may be an additional target for prophylactic intervention.
Lung
Acute chest syndrome (ACS), which is defined as a new infiltrate on chest radiograph associated with fever and respiratory symptoms (cough, wheezing, and tachypnea), is a leading cause for hospital admissions and deaths in SCD.13 Studies estimate that as many as 90% of adults with SCD have abnormal lung function.14,15 The pathophysiology is complex and includes vasoocclusion due to polymerization of sickle hemoglobin in the relatively hypoxic environment of pulmonary capillaries, hypoxia-related vasoconstriction, release of inflammatory cytokines in response to infection or fat embolism, and endothelial activation with enhanced RBC adhesion.16,17 Restrictive lung disease in adults with SCD is presumed to result from repeated insults during childhood, including ACS.18 Chronic hypoxemia and pulmonary hypertension are additional components of sickle lung disease in adults, however, there are limited data on how they relate to early changes in lung function or structure in young children.
Several longitudinal and cross-sectional studies of lung function in children have demonstrated a progressive decline in lung volumes with early lower airway obstruction, restriction, and airway hyperreactivity.19–22 The bronchodilator response and patterns of cytokine expression in SCD share features with asthma, and suggest that wheezing and obstruction may be manifestations of SCD or of concurrent comorbid states.23 Comprehensive assessment of lung disease typically includes spirometric pulmonary function testing that may not be feasible in younger children. Evaluation of preschool children 2-5 years of age with cystic fibrosis and age-matched normal controls suggest that very young children are capable of completing spirometric studies with motivated, experienced staff.24 To date, there are no published pulmonary function studies in a comparable young SCD cohort. Passive occlusion and nitrogen washout techniques under sedation have been used to obtain limited pulmonary function data on a small, uncontrolled cohort of infants as young as 3 months of age with SCD, and demonstrated findings suggestive of lower airway obstruction and hyperreactivity in some infants.25 It is not clear that this approach is feasible in terms of safety for larger-scale studies or clinical practice, so these data have not yet been reproduced. The feasibility of designing studies to directly address primary prophylaxis for pulmonary complications will require further investigations to identify diagnostic modalities that can be used in the presymptomatic state, likely in very early childhood.
Brain
Cerebrovascular events such as overt strokes occur in 24% of individuals with SCD by the age of 45 years.26 The impact of SCD on the brain has other manifestations as well, including silent cerebral infarcts, altered cerebral vascular flow velocity, and impaired neurocognitive function. The pathologic findings in most but not all cases of overt stroke consist of intimal thickening in medium to large intracranial blood vessels likely due to recurrent sickling-induced endothelial injury with thrombosis.27 Silent cerebral infarcts, high-signal MRI abnormalities in the absence of overt neurological signs, are detectable in 20%-35% of children28 and are a risk factor for overt stroke and cognitive impairment. Studies performed on infants at the initiation of the BABY HUG study identified silent cerebral infarcts in 13% at a median age of 13.7 months, suggesting that the brain injury represented by these lesions occurs at a very early age.29 Altered cerebral blood flow due to severe anemia and/or stenosis may be manifested by elevated flow velocity shown by transcranial Doppler (TCD). Approximately 10% of children in the Stroke Prevention Trial in Sickle Cell Anemia (STOP) had velocities in the abnormal range, most often in children 2-8 years of age.30 Baseline data from the BABY HUG trial identified no patients at a mean age of 12.6 months with abnormal TCDs,31 providing a potential window of opportunity for early intervention.
Primary prophylactic interventions in SCD
Penicillin
The efficacy of oral penicillin as primary prophylaxis in SCD was clearly demonstrated in pivotal trials published nearly 25 years ago.32,33 In a study by Gaston et al, children who were randomized to the daily oral penicillin prophylaxis arm of the study had an 85% reduction in the probability of infection compared with those given placebo. This study provided much-needed impetus for states in the United States to adopt universal newborn testing to ensure that all children with SCD were identified, with a strong recommendation for prompt penicillin initiation.34 The clear benefit of newborn screening combined with penicillin prophylaxis is demonstrated by decreases in SCD-related mortality among black children under the age of 4 years by 68% from 1983-1986 to 1999-2002, with the largest decrease occurring after 2000, when the 7-valent pneumococcal vaccine was added to the schedule of recommended immunizations for all children.35,36 Similar improvements in survival were noted in the Dallas Newborn Cohort, in which all children identified through newborn screening were immunized, given penicillin prophylaxis at least through the age of 5 years, and provided with comprehensive medical care.37,38 The proportion of children surviving to age 18 years was 93.9%, and sepsis was no longer the leading cause of deaths.
A follow-up study to determine acceptable circumstances for discontinuation of penicillin was restricted to fully immunized children with homozygous sickle cell and sickle beta zero thalassemia who had been compliant with penicillin, had no serious infections in the first 5 years of life, and were not splenectomized.39 Episodes of pneumococcal infections occurred at lower rates than anticipated in both arms of this randomized, double-blind, placebo-controlled study. Because no significant difference could be detected, the investigators concluded that penicillin prophylaxis could be safely discontinued at age 5 years, although some pediatric hematologists continue to recommend indefinite prophylaxis given concerns about infection susceptibility.40 Subsequent studies examining invasive pneumococcal infections at regional pediatric SCD programs in the era of penicillin prophylaxis, immunization with the 23-valent pneumococcal vaccine in children with SCD, and the addition of the 7-valent–conjugated pneumococcal vaccine to the standard schedule in all children have demonstrated benefits in survival and infection rates.41,42
Hydroxyurea
Hydroxyurea is a well-characterized pharmacologic agent initially used as an antineoplastic drug that inhibited ribonucleotide reductase in DNA synthesis. Because hydroxyurea use was associated with an increase in HbF, and mechanistically because endogenous or induced fetal hemoglobin elevation is known to reduce the severity of SCD, hydroxyurea became a focus of translational and clinical research in SCD. Hydroxyurea-induced marrow suppression favors proliferation of RBC precursors containing HbF. Overall hemoglobin content is increased and hydroxyurea improves sickle RBC hydration and reduces RBC adherence to endothelial cells. The chronology of pivotal clinical trials of hydroxyurea and related observations was eloquently summarized by Ware and Aygun.43 The phase 3 randomized Multicenter Study of Hydroxyurea (MSH) trial in adults with SCD established the efficacy of hydroxyurea in reducing pain severity and frequency of hospitalizations for pain management, and also showed a decrease in ACS among participants taking hydroxyurea.44 A small open-label pilot study of severely affected children with SCD suggested that hydroxyurea was associated with a reduction in the length and frequency of admissions.45 These observations were extended to severely affected children 5-15 years of age in the phase 1/2 HUG-KIDS study, which demonstrated both safety and efficacy in this age cohort.46 A systematic review of hydroxyurea in children with SCD was commissioned by the National Institutes of Health Office of Medical Applications of Research to determine the strength of evidence on its efficacy, toxicity, and effectiveness.47 Whereas the efficacy of hydroxyurea to induce fetal hemoglobin and reduce hospital admissions was strong, evidence of its benefit in preserving splenic function was considered low.
The impact of hydroxyurea on the prevention of microalbuminuria or the progression of renal dysfunction in asymptomatic children has been suggested by some small observational studies.5,48 In general, children without microalbuminuria who were treated with hydroxyurea for nonrenal clinical manifestations of SCD did not develop microalbuminuria, and some who had microalbuminuria when hydroxyurea was initiated did not worsen. The combined use of hydroxyurea with an ACE inhibitor such as enalapril resulted in reduced urinary protein excretion and improved creatinine clearance.
Reports on the capacity of hydroxyurea to impact splenic function in SCD in severely affected older children have shown variable responses.49–52 The first clinical trial designed specifically to study hydroxyurea for primary prophylaxis in SCD came with the Hydroxyurea Safety and Organ Toxicity (HU-SOFT) trial.53 This prospective, open-label, multicenter study sought to determine the feasibility of administering hydroxyurea to very young children (median age 15 months) while assessing toxicity, hematologic responses, and retention of splenic function. Hydroxyurea was well tolerated and had acceptable short-term toxicity in this fixed-dose, 2-year study. Among children with liver-spleen scans performed at both study entry and exit, most had unchanged or reduced splenic uptake over time. Splenic uptake was absent at the end of the study period for 47% of HU-SOFT participants, which compared favorably to children in the Cooperative Study of Sickle Cell Disease, in which 78% of children had pitted RBC counts > 3.5% by the age of 3 years.54 An extended study with dose escalation continued to demonstrate hematologic responses with increased fetal hemoglobin, normal growth, and no novel toxicities.55 In examining those subjects with serial assessments of splenic function, 21.5% had normal uptake after a mean duration of 4.9 years on hydroxyurea, whereas 43% at extension exit had absent splenic uptake, at an age when the majority of children with homozygous sickle cell historically were presumed to have functional asplenia.
These data created a strong scientific basis for the design of the BABY HUG trial. In this multicenter, double-blind, placebo-controlled trial, 193 young children, 9-18 months of age with homozygous sickle cell or sickle beta zero thalassemia were randomized to receive or not receive hydroxyurea at a fixed dose. The primary end points for this study were reduction in the decline of splenic function measured by liver-spleen scan by 50% with hydroxyurea and attenuation of the increase in glomerular filtration based on 99Tc-DTPA clearance.56 Secondary end points included comparison of liver spleen scans with other laboratory assessments of splenic function, the utility of calculated estimates of GFR for determining renal function, and investigations of growth, neurodevelopment, and mutagenesis. Adverse events, including expected SCD manifestations, potential treatment toxicities, and serious unanticipated events, were also evaluated.
With greater numbers of infants at study entry having diminished splenic uptake and altered glomerular filtration than reported in previous studies, BABY HUG did not detect significant improvements over the study period in either renal or splenic function with hydroxyurea compared with placebo, and therefore the study failed to meet the primary study end points.57 There are several potential explanations for these findings. As with previous studies on hydroxyurea in very young children, a fixed dose schedule (20 mg/kg) was used, presumably to abrogate toxicity. Greater impact on organ function might have been achieved had the study design allowed for dose escalation. Furthermore, the decline in splenic function in the placebo group was smaller than expected, and therefore the relative difference with the hydroxyurea may have been less significant. It is unclear if the manner in which the assays were used, such as the qualitative interpretation of the splenic uptake, adversely affected the study outcome or if there is in fact no true benefit to hydroxyurea prophylaxis in this setting. In prior studies, interpretation of scintigraphic studies (“normal” versus “present but reduced”) may have not been comparable to BABY HUG, creating study design challenges. The duration of the core study may not have been adequate to detect significant changes, and the imposition of a 3-month clinical hold on drug distribution may have also had some effect. It is also conceivable that with a longer observation period, as proposed in the BABY HUG follow-up, open-label extension study, other clinical benefits will accrue.
Despite the limitations of BABY HUG to demonstrate direct benefit of hydroxyurea as primary prophylaxis, the study makes important contributions to our understanding of the pathophysiology of early disease manifestations, which might be further explored in future studies. As secondary end points, the study found reductions in the frequency of pain episodes and ACS and fewer hospitalizations for subjects who received hydroxyurea compared with controls, findings that have been associated with reduced early mortality in other SCD studies. As seen in most studies of hydroxyurea in SCD, there were significant changes in laboratory parameters, such as increased total hemoglobin and fetal hemoglobin and lower WBC counts in participants who received hydroxyurea on BABY HUG, without excess or novel toxicities. The relationship of these findings to improved long-term outcomes, including reduced mortality, has been demonstrated in other SCD trials,55,58 but this is beyond the stated objectives of the BABY HUG study.
Transfusion
Relief of anemia, relative reductions in the percentage of circulating RBCs, high intracellular concentrations of sickle hemoglobin, and relative improvements in oxygen-carrying capacity are the most common goals for acute or chronic RBC transfusions. Chronic transfusions at present are the most effective intervention to prevent recurrent cerebrovascular events in SCD patients who have had overt strokes. Transfusion for primary prevention in children with SCD and elevated blood flow velocities in major cerebral blood vessels was demonstrated in the STOP trial.30 This study demonstrated a 90% reduction in risk of stroke in the transfusion group, leading to the recommendation that all SCD children 2-16 years of age have an annual screening TCD, and that chronic transfusions be initiated in those with documented abnormal velocities. The effectiveness of TCD screening and primary intervention has now been demonstrated by 2 large independent comprehensive SCD programs, with significant reductions (3- to 10-fold) in overt stroke incidences since starting routine screening with TCD.59,60
Can predictors of disease severity identify additional targets for prophylaxis?
SCD complications among the infant cohort from the CCSCD were examined to create a model to predict adverse outcomes by the age of 10 years.61 Children with severe anemia (hemoglobin < 7 g/dL), early dactylitis (< 1 year of age), and leukocytosis were more likely to have severe disease manifestations such as stroke, recurrent pain, or ACS. The presence of > 3.5% pocked RBCs at 1 year of age was also a significant predictor of adverse outcomes, but was not available for all participants. This model was validated retrospectively; however, a more recent application of these predictors to an independent cohort could not reproduce these findings.62
New biomarkers of kidney injury may be useful in predicting sickle nephropathy. Urine kidney injury molecule 1 (KIM-1) and N-acetyl-b-D-glucosaminidase (NAG) are strongly correlated with increasing microalbuminuria and proteinuria with age in SCD.63 Serum cystatin C, which has been used as a marker of renal dysfunction in a variety of clinical conditions, is inversely correlated with DTPA GFR, and thus may be an early marker of SCD-related hyperfiltration.64
Gene-association studies may be useful in focusing on certain aspects of disease pathophysiology that might be exploited in new primary prevention strategies, or they may aid in the prediction of which patients are likely to respond to an intervention. The phenotypic heterogeneity of SCD is likely the result of modifying genes. The best examples of genetic modifiers in SCD are regulators of fetal hemoglobin production and coinheritance of alpha thalassemia.65,66 The XmnI polymorphism in the gamma globin promoter, the HBS1-MYB intergenic sequences, and BCL11A account for 20%-50% of the variations in fetal hemoglobin levels in SCD,67 and may be targets for new therapies in the future and potential markers to incorporate into the design of future prophylaxis studies.
Conclusion
Penicillin prophylaxis and transfusions in children at high risk of strokes are prime examples of the feasibility and effectiveness of early intervention. Primary prevention as a strategy to reduce morbidity and mortality due to other SCD-related complications remains elusive. Recent studies continue to define the natural history of SCD and new opportunities for intervention. Enhanced knowledge of disease pathophysiology and the development of clinical tools to accurately assess early manifestations of organ dysfunction are of vital importance and currently limit the feasibility of interventions specifically focused on primary prevention. The correlation of splenic biomarkers with absent splenic function on liver-spleen scans in very young children with SCD provides a framework for future studies. More longitudinal studies with validated tools to assess renal and pulmonary changes over time in SCD are needed to link manifestations during childhood to later progressive organ injury. Hydroxyurea continues to show benefit in reducing pain and ACS across the lifespan, and its more widespread use for these indications should be pursued; however, its role in preventing organ dysfunction needs further investigation. Ongoing engagement of researchers and the SCD community to launch and successfully complete clinical trials will be essential. Opportunities for future studies that focus on the prevention of SCD complications may ultimately improve survival and quality of life for these patients.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Hydroxyurea is FDA-approved for use only in adult SCD patients.
Correspondence
Alexis A. Thompson, MD, MPH, Division of Hematology, Oncology & Stem Cell Transplant, Children's Memorial Hospital, Box 30, 2300 Children's Plaza, Chicago, IL 60614; Phone: (773) 880-4562; Fax: (773) 880-3223; e-mail: a-thompson@northwestern.edu.
