
Abstract
Platelets have evolved highly specialized adhesion mechanisms that enable cell-matrix and cell-cell interactions throughout the entire vasculature irrespective of the prevailing hemodynamic conditions. This unique property of platelets is critical for their ability to arrest bleeding and promote vessel repair. Platelet adhesion under conditions of high shear stress, as occurs in stenotic atherosclerotic arteries, is central to the development of arterial thrombosis; therefore, precise control of platelet adhesion must occur to maintain blood fluidity and to prevent thrombotic or hemorrhagic complications. Whereas the central role of platelets in hemostasis and thrombosis has long been recognized and well defined, there is now a major body of evidence supporting an important proinflammatory function for platelets that is linked to host defense and a variety of autoimmune and inflammatory diseases. In the context of the vasculature, experimental evidence indicates that the proinflammatory function of platelets can regulate various aspects of the atherosclerotic process, including its initiation and propagation. The mechanisms underlying the proatherogenic function of platelets are increasingly well defined and involve specific adhesive interactions between platelets and endothelial cells at atherosclerotic-prone sites, leading to the enhanced recruitment and activation of leukocytes. Through the release of chemokines, proinflammatory molecules, and other biological response modulators, the interaction among platelets, endothelial cells, and leukocytes establishes a localized inflammatory response that accelerates atherosclerosis. These inflammatory processes typically occur in regions of the vasculature experiencing low shear and perturbed blood flow, a permissive environment for leukocyte-platelet and leukocyte-endothelial interactions. Therefore, the concept has emerged that platelets are a central element of the atherothrombotic process and that future therapeutic strategies to combat this disease need to take into consideration both the prothrombotic and proinflammatory function of platelets.
Introduction
Platelets are anuclear cell fragments derived from the cytoplasm of BM megakaryocytes1 that circulate in the bloodstream of humans for 7-10 days. The primary hemostatic function of platelets has been recognized for more than a century, and the molecular processes regulating the adhesive function of platelets have been elucidated in great detail.2–4 In addition to preventing excess bleeding, platelets also play an important role in surveying and maintaining the integrity of the endothelium, in part through the release of proangiogenic cytokines and growth factors. Dysregulated platelet-endothelial interactions have increasingly been recognized as an important pathogenic mechanism in the development of atherosclerosis. In response to specific proinflammatory signals, endothelial cells become more adhesive toward platelets, stimulating the production of various platelet-derived inflammatory molecules that provide a positive feedback loop for further endothelial cell activation. Endothelial-bound platelets are highly effective at recruiting leukocytes from flowing blood and also enhance leukocyte adhesion and transmigration to the site of the proinflammatory stimulus. Therefore, pathological derangement of these key interactions among platelets, endothelial cells, and leukocytes facilitates the inflammatory process that underlies the development of atherosclerosis.
This review outlines the key pathways used by platelets to facilitate the development of vascular inflammatory lesions linked to the development of atherosclerosis, with a focus on the mechanisms leading to dysregulation of the platelet-endothelium and platelet-leukocyte interaction. The importance of local blood flow disturbances in regulating the atherothrombotic process is briefly discussed. A brief overview of the well-defined platelet adhesion mechanisms regulating platelet-vessel wall and platelet-platelet interactions and a discussion of recent advances in the understanding of the platelet procoagulant response are presented first.
Hemostatic and prothrombotic function of platelets
Under normal physiological conditions, platelets circulate in close proximity to the endothelium without forming stable adhesion contacts due to the anti-adhesive properties of quiescent endothelial cells. However, after vascular injury, platelets rapidly adhere to sites of endothelial disruption to establish a hemostatic plug that prevents excess blood loss. This process is critically dependent on the efficiency of platelet adhesion to the subendothelial matrix, as well as on the ability of the platelets to undergo rapid biochemical and morphological changes that support aggregation and the localized activation of the coagulation cascade.
Platelet recruitment (Figure 1)

Hemostatic and prothrombotic function of platelets under flow. (A) Platelet adhesion mechanisms operating under arterial flow. After disruption of the endothelium, platelets are rapidly recruited from flowing blood via a tethering mechanism dependent on the interaction of platelet GPIb-V-IX and VWF. This adhesive bond has a rapid dissociation rate, resulting in platelet translocation on the vessel wall. (B) Platelet firm adhesion and aggregation. Translocating platelets engage collagen in the vessel wall through their adhesion receptors GPVI and α2β1. GPVI is the major collagen receptor inducing intracellular calcium flux necessary for stable platelet adhesion, cytoskeletal reorganization, αIIbβ3 activation, and the release of the soluble agonists (ADP and TxA2). These agonists act through specific G-protein–coupled receptors to amplify the platelet activation response. TxA2 is derived from the metabolism of arachidonic acid via the COX-1 pathway, whereas ADP is released from platelet-dense granules. Locally generated thrombin also enhances platelet activation through proteolytic cleavage and activation of PARs. Activated platelets form stable aggregates through αIIbβ3 engagement of VWF and fibrinogen. These molecular pathways of amplification of platelet activation are the target of both clinically available and experimental therapeutic interventions in the treatment of atherothrombotic disease. (C) Stabilization of the platelet thrombus. The primary hemostatic plug is consolidated by fibrin generation at the site of injury and throughout the developing thrombus. α-Thrombin generation at the injured vessel wall is critically dependent on TF, whereas thrombin generation on the surface of activated platelets is likely to evolve both TF and contact factor–dependent activation of blood coagulation. UHF indicates unfractionated heparin; LMWH, low-molecular-weight heparin; VKA, vitamin K antagonist; PGH2, prostaglandin H2; AA, arachidonic acid, PLA2, phospholipase A2; IP3R, inositol trisphosphate receptor; ITAM, immunoreceptor tyrosine-based activation motif; FcRγ, Fc receptor gamma.
Hemostatic and prothrombotic function of platelets under flow. (A) Platelet adhesion mechanisms operating under arterial flow. After disruption of the endothelium, platelets are rapidly recruited from flowing blood via a tethering mechanism dependent on the interaction of platelet GPIb-V-IX and VWF. This adhesive bond has a rapid dissociation rate, resulting in platelet translocation on the vessel wall. (B) Platelet firm adhesion and aggregation. Translocating platelets engage collagen in the vessel wall through their adhesion receptors GPVI and α2β1. GPVI is the major collagen receptor inducing intracellular calcium flux necessary for stable platelet adhesion, cytoskeletal reorganization, αIIbβ3 activation, and the release of the soluble agonists (ADP and TxA2). These agonists act through specific G-protein–coupled receptors to amplify the platelet activation response. TxA2 is derived from the metabolism of arachidonic acid via the COX-1 pathway, whereas ADP is released from platelet-dense granules. Locally generated thrombin also enhances platelet activation through proteolytic cleavage and activation of PARs. Activated platelets form stable aggregates through αIIbβ3 engagement of VWF and fibrinogen. These molecular pathways of amplification of platelet activation are the target of both clinically available and experimental therapeutic interventions in the treatment of atherothrombotic disease. (C) Stabilization of the platelet thrombus. The primary hemostatic plug is consolidated by fibrin generation at the site of injury and throughout the developing thrombus. α-Thrombin generation at the injured vessel wall is critically dependent on TF, whereas thrombin generation on the surface of activated platelets is likely to evolve both TF and contact factor–dependent activation of blood coagulation. UHF indicates unfractionated heparin; LMWH, low-molecular-weight heparin; VKA, vitamin K antagonist; PGH2, prostaglandin H2; AA, arachidonic acid, PLA2, phospholipase A2; IP3R, inositol trisphosphate receptor; ITAM, immunoreceptor tyrosine-based activation motif; FcRγ, Fc receptor gamma.
The mechanisms supporting platelet adhesion to the damaged vessel wall have been thoroughly investigated and well defined, so only a brief description will be provided here. After damage to the vessel wall, subendothelial matrix proteins that are highly reactive to platelets, including collagen, VWF, fibronectin, and laminin, become exposed to the blood and immediately engage specific receptors on the platelet surface. The contribution of specific ligands and receptors mediating platelet adhesion is critically dependent on the prevailing blood-flow conditions. Under conditions of high shear stress, as encountered in arterioles and stenotic arteries, VWF plays a critical role in recruiting platelets from blood flow.5 This tethering function of subendothelial VWF is dependent on the interaction of its A1 domain with the glycoprotein Ibα (GPIbα) component of the platelet GPIb-V-IX complex.6 Circulating plasma VWF has limited binding potential for GPIbα, however, once immobilized onto subendothelial collagen (type I, III, and VI), the unfolded VWF macromolecule provides a linear array of A1 domains that facilitate binding to multiple GPIbα receptors.7,8 The bond between GPIbα and VWF has a rapid dissociation rate that is unable to support stable adhesion, requiring the contribution of other ligand-receptor interactions to promote stable adhesion.9 The fundamental importance of both VWF and GPIbα for the adhesive function of platelets is underscored by the severe bleeding disorder experienced by patients with qualitative or quantitative defects in these molecules.10
Firm adhesion and activation (Figure 1)
Once tethered to the site of vascular injury, platelets form stable adhesion contacts with collagen and other matrix macromolecules. Platelet binding to collagen is mediated by GPVI11,12 and integrin α2β1,13 whereas fibronectin and laminin engage integrin α5β1 and α6β1, respectively.4 Isolated deficiency of any of these receptors does not cause a severe bleeding disorder, suggesting considerable redundancy in the mechanisms regulating platelet-vessel wall interactions. Once adherent, platelets undergo a remarkably complex series of morphological and biochemical changes that lead to the release of platelet granule contents and up-regulation of the adhesive function of integrin αIIbβ3. Activated αIIbβ3 binds multiple ligands, including VWF,14,15 fibrinogen,16 fibrin and fibronectin,17 and is indispensable for the formation of stable platelet aggregates.18 Therefore, similar to GPIb, quantitative or qualitative defects in αIIbβ3 leads to a major defect in the hemostatic function of platelets and a severe bleeding disorder (Glanzmann thrombasthenia).19 Integrin αIIbβ3 deficiency also provides protection against arterial thrombosis, thus pharmacological αIIbβ3 antagonism represents a highly effective antithrombotic approach.20,21
Soluble agonists amplifying platelet activation (Figure 1)
Central to platelet activation is the generation and release of soluble agonists at sites of vascular injury. These include thromboxane A2 (TxA2) which is synthesized from arachidonic acid via cyclooxygenase (COX) and thromboxane synthase, and ADP released from platelet dense granules. These endogenous agonists can act in an autocrine or paracrine manner to enhance platelet activation by engaging specific G protein coupled receptors; P2Y122 and P2Y1223 (ADP) and the thromboxane receptors TPα and TPβ.24 ADP and TxA2 act in a cooperative manner to enhance platelet activation and thrombus formation, thus pharmacological targeting of TxA225 generation and/or the P2Y1226 receptor are effective strategies to reduce thrombus propagation at sites of vascular injury.27,28
Thrombin (Figure 1)
In addition to the synthesis and release of soluble agonists, platelets provide a catalytic surface for the assembly of coagulation complexes necessary for α-thrombin generation. Thrombin is among the most potent stimulators of platelets through proteolytic cleavage and activation of platelet protease-activated receptors (PARs), specifically PAR1 and PAR4, on human platelets.29 Also central to the hemostatic function of thrombin is its ability to generate fibrin polymers, a key step stabilizing formed thrombi. Therefore, pharmacological inhibitors of thrombin (eg, direct thrombin inhibitors), upstream coagulation proteases (vitamin K antagonists and factor Xa [FXa] inhibitors), and, potentially, thrombin receptors (PAR1 antagonists), represent effective strategies to limit arterial thrombus growth.27
Procoagulant function of platelets: emerging concepts
Current therapeutic strategies aimed at reducing thrombin's effects in vivo primarily target various components of the coagulation cascade, thus affecting thrombin generation at the vessel wall as well as within the body of thrombus. However, recent progress in understanding the mechanisms by which platelets support blood coagulation have raised the possibility that selectively inhibiting platelet procoagulant function may provide a targeted approach to specifically reduce thrombin generation within the thrombus.
Platelet regulation of the contact phase of blood coagulation (Figure 2)

Procoagulant function of platelets. (A) Plasma membrane phospholipids are asymmetrically distributed in resting platelets, with PS confined to the inner leaflet. After platelet activation by potent agonists, high levels of sustained intracellular calcium triggers scramblase activity. Scramblase activation results in the loss of phospholipid asymmetry and PS exposure on the outer leaflet of the platelet membrane, providing the requisite surface for rapid thrombin generation. (B) PS exposure can be induced through two distinct cell-death pathways; apoptosis and necrosis. Apoptosis results in assembly of the Bak/Bax mitochondrial membrane pore, causing release of cytochrome C (CytC), caspase activation, and substrate proteolysis. Necrotic cell death can be initiated by elevated cytosolic Ca2+, causing a loss in mitochondrial membrane potential and subsequent formation of a mitochondrial permeability transition pore (mPTP). mPTP formation results in bioenergetic failure through ATP depletion with consequent reactive oxygen species (ROS) generation, leading to loss of membrane integrity. (C) Platelets can also contribute to thrombin generation via activation of FXII through the release of polyphosphate from dense granules. (D) Whether there are additional cell death pathways regulating PS exposure and the procoagulant function of platelets remains unclear. PC indicates phosphatidylcholine.
Procoagulant function of platelets. (A) Plasma membrane phospholipids are asymmetrically distributed in resting platelets, with PS confined to the inner leaflet. After platelet activation by potent agonists, high levels of sustained intracellular calcium triggers scramblase activity. Scramblase activation results in the loss of phospholipid asymmetry and PS exposure on the outer leaflet of the platelet membrane, providing the requisite surface for rapid thrombin generation. (B) PS exposure can be induced through two distinct cell-death pathways; apoptosis and necrosis. Apoptosis results in assembly of the Bak/Bax mitochondrial membrane pore, causing release of cytochrome C (CytC), caspase activation, and substrate proteolysis. Necrotic cell death can be initiated by elevated cytosolic Ca2+, causing a loss in mitochondrial membrane potential and subsequent formation of a mitochondrial permeability transition pore (mPTP). mPTP formation results in bioenergetic failure through ATP depletion with consequent reactive oxygen species (ROS) generation, leading to loss of membrane integrity. (C) Platelets can also contribute to thrombin generation via activation of FXII through the release of polyphosphate from dense granules. (D) Whether there are additional cell death pathways regulating PS exposure and the procoagulant function of platelets remains unclear. PC indicates phosphatidylcholine.
The generation of thrombin at the injured vessel wall is contingent upon the expression of tissue factor (TF) on the surface of fibroblasts, smooth muscle cells, and potentially endothelial cells and leukocytes. TF expression and activation helps to stabilize the active conformation of FVIIa,30 triggering the extrinsic pathway of blood coagulation. The mechanisms underlying the generation of thrombin within the body of a developing thrombus remains controversial. Whereas TF is expressed on the surface of activated leukocytes and microparticles and has been demonstrated to be incorporated within the thrombus, recent experimental evidence has suggested a potentially important role for the contact phase of blood coagulation in promoting arterial thrombus growth. This rekindled interest in the role of contact factors (eg, FXII, FXI, and high-molecular-weight kininogen) in promoting blood coagulation in vivo stemmed from the observation that FXII-deficient mice are protected from vasoocclusive thrombi in arterial thrombosis models.31 Similarly, FXI deficiency32,33 and pharmacological antagonism34 of the contact phase of coagulation leads to a similar defect in arterial thrombosis. Given that humans with complete FXII deficiency do not have a significant hemostatic defect, this has raised the possibility that therapeutic targeting of this process may lead to the development of safer antithrombotics. How platelets activate the contact phase of coagulation during thrombus development has remained a vexing issue.35 Recently, Muller et al identified inorganic polyphosphate released from activated platelets as a major regulator of FXII activation.36 Pharmacologic exploitation of this pathway may enable targeting of platelet polyphosphate by phosphatases to reduce thrombotic risk while leaving hemostatic responses intact.
Identification of the elusive phospholipid scramblase
The contribution of platelet-derived phospholipids to thrombin generation has long been recognized; however, the biological mechanisms regulating this procoagulant platelet function have remained poorly defined. In resting platelets, membrane phospholipids are asymmetrically distributed: phosphatidylcholine and sphingomyelin are primarily localized in the outer leaflet of the plasma membrane, whereas phosphatidylethanolamine and phosphatidylserine (PS) are largely confined to the inner leaflet. This asymmetry is maintained through the action of two membrane lipid transporters, flippase and floppase.37,38 After platelet stimulation by potent agonists, this asymmetry is lost due to activation of Ca2+-dependent phospholipid scramblases that bidirectionally transport phospholipids.39 Scramblase activity therefore results in the expression of negatively charged phospholipids, especially PS, on the outer leaflet of the platelet membrane, thus providing the requisite surface for assembly of coagulation factors required for thrombin generation (Figure 2). Defects in platelet PS expression are rare; however, in affected individuals, this leads to a major hemostatic disturbance, as described in Scott syndrome.40 Until recently, the identity of the phospholipid scramblase had remained elusive, but recent studies have identified a plasma membrane protein transmembrane protein 16F (TMEM16F) that confers Ca2+-dependent scrambling of phospholipids. A mutation in the TMEM16F gene resulting in premature termination of the protein has been demonstrated in a patient with Scott syndrome.41
Cell death and the procoagulant function of platelets (Figure 2)
Procoagulant platelets expressing PS exhibit distinct morphologic and biochemical features relative to fully activated platelets. Many of these changes are indicative of dead or dying cells, and include membrane blebbing, microvesiculation, activation of intracellular proteases, and the cleavage of cytoskeletal proteins, PS surface exposure, and alterations in mitochondrial membrane potential.42 These results suggest that the pathways regulating cell death in platelets may play an important role in regulating the platelet procoagulant phenotype. Programmed cell death is orchestrated by several pathways, including apoptosis, necrosis, and autophagy. Apoptosis is an energy-dependent process contingent upon caspase activation and is critical for the removal of senescent cells. Necrosis is typified by bioenergetic failure of the cell resulting in membrane instability and the release of intracellular contents into the extracellular environment. Recently, evidence has emerged that platelet stimulation by potent agonists initiates a tightly regulated cell death pathway mediated by sustained high levels of cytosolic Ca2+, culminating in distinct morphological and biochemical changes that are consistent with a necrotic cell death pathway.43 Direct induction of apoptosis can also induce PS expression and platelet procoagulant function,43 raising the possibility that multiple cell death pathways may regulate thrombin generation on the surface of platelets. In addition to promoting thrombin generation, procoagulant platelets may also contribute to the inflammatory response by generating high levels of the proinflammatory lipid platelet-activating factor (PAF) and by enhancing leukocyte adhesion.44
Proinflammatory role of platelets
Promiscuous platelets
The growing list of pathophysiological processes in which platelets have a proposed role is in part a reflection of the large number of different cell types with which platelets can interact. These include endothelial cells, neutrophils, monocytes, dendritic cells, cytotoxic T-lymphocytes, malaria-infected red blood cells, and various tumor cells. In the context of atherosclerosis, the interaction of platelets with endothelial cells and leukocytes appears to be important for the initiation and propagation of inflammatory processes in the arterial wall.45
Platelet-endothelial cell adhesive interactions (Figure 3)

Platelet-endothelial cell interactions. (A) The healthy endothelium. The anti-adhesive phenotype of endothelial cells is maintained through 3 intrinsic pathways: the ecto-ADPase/CD39/NTPDase pathway, which metabolizes ADP, and the PGI2 and NO pathways, which inhibit platelet activation through the stimulation of cAMP and cGMP production, respectively. (B) Endothelial activation in hyperlipemia. In a hyperlipidemic milieu, the endothelium becomes inflamed as a result of modified lipoprotein particles and reactive oxygen species (ROS) accumulating in the intima. This leads to the expression of adhesive ligands (VWF and P- and E-selectins) on the endothelium. (C) Platelet adhesion to the inflamed endothelium and platelet endothelial cross-talk. The expression of VWF and P-selectin on the endothelial surface supports platelet tethering and rolling. Subsequently, stable platelet adhesion occurs through fibrinogen-αIIbβ3 complexes binding to αvβ3 or ICAM-1 on endothelial cells. Adherent platelets secrete numerous bioactive substances that alter the chemotactic and adhesive properties of the endothelial cells. Platelet-derived IL-1β induces endothelial secretion of IL-6 and IL-8 and surface expression of ICAM-1, αvβ3, and MCP-1. Platelet CD40 ligand binds CD40 on endothelial cells, resulting in up-regulation of adhesive molecules (ICAM-1, VCAM-1, and E- and P-selectin), cytokine and TF release, and reduction in NO synthesis. Ecto ADPase indicates ecto-adenosine diphosphatase; NTPDase, nucleoside triphosphate diphosphohydrolase.
Platelet-endothelial cell interactions. (A) The healthy endothelium. The anti-adhesive phenotype of endothelial cells is maintained through 3 intrinsic pathways: the ecto-ADPase/CD39/NTPDase pathway, which metabolizes ADP, and the PGI2 and NO pathways, which inhibit platelet activation through the stimulation of cAMP and cGMP production, respectively. (B) Endothelial activation in hyperlipemia. In a hyperlipidemic milieu, the endothelium becomes inflamed as a result of modified lipoprotein particles and reactive oxygen species (ROS) accumulating in the intima. This leads to the expression of adhesive ligands (VWF and P- and E-selectins) on the endothelium. (C) Platelet adhesion to the inflamed endothelium and platelet endothelial cross-talk. The expression of VWF and P-selectin on the endothelial surface supports platelet tethering and rolling. Subsequently, stable platelet adhesion occurs through fibrinogen-αIIbβ3 complexes binding to αvβ3 or ICAM-1 on endothelial cells. Adherent platelets secrete numerous bioactive substances that alter the chemotactic and adhesive properties of the endothelial cells. Platelet-derived IL-1β induces endothelial secretion of IL-6 and IL-8 and surface expression of ICAM-1, αvβ3, and MCP-1. Platelet CD40 ligand binds CD40 on endothelial cells, resulting in up-regulation of adhesive molecules (ICAM-1, VCAM-1, and E- and P-selectin), cytokine and TF release, and reduction in NO synthesis. Ecto ADPase indicates ecto-adenosine diphosphatase; NTPDase, nucleoside triphosphate diphosphohydrolase.
While platelets continuously circulate in close proximity to the endothelium, under physiological conditions, they typically do not form sustained adhesive interactions. The anti-adhesive phenotype of endothelial cells is controlled by the three intrinsic pathways; the nitric oxide (NO) pathway, the ecto-ADPase/CD39/NTPDase pathway, and the ecosanoid-arachidonic acid-prostacyclin (PGI2) pathway.46 It is increasingly recognized that inflammatory stimuli facilitate sustained endothelial-platelet adhesion by perturbing these anti-adhesive mechanisms and increasing surface expression of endothelial adhesion molecules.47 The adhesion of platelets to “inflamed endothelium” involves a similar coordinated multistep process as occurs in hemostasis and thrombosis, including platelet tethering, surface translocation, and firm adhesion.48 Initiation of the inflammatory insult to the endothelium in atherosclerosis is partly due to the accumulation of modified oxidized lipoprotein particles in the setting of hyperlipidemia.49 This leads to surface expression of endothelial selectins, including P- and E-selectin, as well as membrane expression of VWF.48,50 The platelet counterreceptors for P-selectin include GPIbα and P-selectin glycoprotein-1 (PSGL-1),51,52 which support tethering and rolling, but not firm adhesion on inflamed endothelium. Whereas there is some uncertainty as to the exact mechanism supporting stable platelet adhesion to activated endothelium, in vitro and in vivo models suggest that binding of αIIbβ3 to fibrinogen bound to endothelial αvβ3 or ICAM-1 facilitates firm platelet adhesion and activation.53,54
Chemical signals
Platelets activated through their interactions with inflamed endothelium release a vast array of inflammatory and mitogenic mediators (Table 1) into the local microenvironment.55 These platelet-derived bioactive substances alter the chemotactic and adhesive properties of the endothelial cells, enabling monocytes and other leukocytes to adhere and transmigrate through the endothelium to sites of inflammation.48,56 In vivo models of atherogenesis suggest that these interactions between endothelial cells and activated platelets are likely to be critical in the initiation of atherosclerosis.57,58
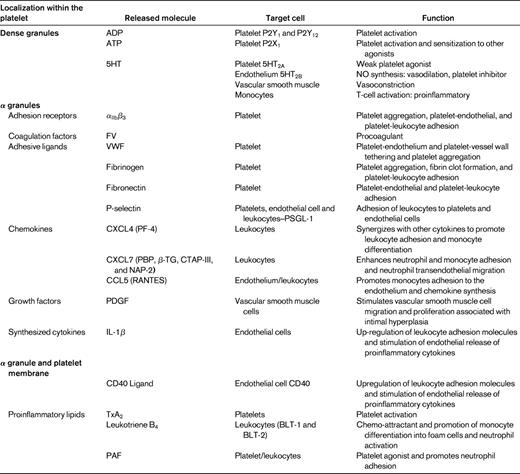
IL-Iβ (Figure 3).
IL-Iβ is a platelet derived cytokine that has been postulated to be a principal mediator of platelet-induced endothelial activation.59,60 It is not stored in platelet granules, but is believed to be synthesized by platelets from constitutional prepackaged mRNA several hours after thrombin stimulation or integrin-mediated adhesion.61 IL-Iβ stimulation of endothelial cells induces secretion of IL-6 and IL-8 and significantly increases the endothelial surface expression of ICAM-1, αvβ3, and monocyte chemotactic protein-1 (MCP-1).48,60,62 ICAM-1 and MCP-1 up-regulation by endothelial cells in response to IL-Iβ is dependent on the activation of NF-κB.63 Through this potential mechanism, platelets are able to significantly enhance monocyte and neutrophil adhesion to the endothelium.
CD40 ligand (CD40L-CD154; Figure 3).
CD40 ligand is an important platelet-derived bioactive mediator of endothelial cell activation. CD40L is a transmembrane protein with structural homology to TNF-α.64 CD40L was initially identified on the surface of activated CD4+ T cells and is central to the function of the humoral immune system by supporting B-cell differentiation and immunoglobulin class switching.65 It was subsequently demonstrated that platelets are a rich source of CD40L,66 with > 95% of circulating CD40L being of platelet origin.67 After release from platelets, CD40L interacts which CD40 expressed on numerous cell types, including endothelial cells, smooth muscle cells, leukocytes, fibroblasts, and platelets.64,68 In endothelial cells, CD40L binding with its cognate receptor CD40 results in up-regulation of ICAM-1, VCAM-1, and E- and P-,selectin as well as the release of IL-6 and TF.69 CD40L inhibits endothelial NO production by destabilizing NO synthase mRNA64,70 ; therefore, CD40L binding to endothelial cells results in a proatherogenic and proinflammatory phenotype.64
Platelet-leukocyte cross-talk in atherogenesis
The central role of leukocytes, especially monocytes and T lymphocytes, in the development of atherosclerosis is well established.71 There is increasing evidence that activated platelets adherent to the inflamed endothelium may enhance leukocyte recruitment, activation, and transmigration, thereby enhancing the inflammatory processes underlying atherosclerosis.
Leukocyte recruitment to endothelial-bound platelets is a coordinated, multistep process involving selectin-mediated tethering and rolling, followed by β2 integrin–mediated stable adhesion (Figure 4). Platelet P-selectin has a central role in mediating the initial capture and rolling of leukocytes through interactions with its key counterreceptor P-selectin glycoprotein 1 (PSGL-1).51,72 Ligation of PSGL-1 induces signaling events culminating in the activation of the leukocyte β2 integrins, including macrophage antigen-1 (Mac-1, αMβ2) and lymphocyte function-associated antigen-1 (LFA-1, αLβ2), which are required to mediate stable leukocyte adhesion. Mac-1 is the principal β2 integrin that facilitates firm adhesion onto the surface of platelets via its binding to multiple ligands, including GPIbα,73 ICAM-2,74 and junctional adhesion molecule-3 (JAM-3),75 as well as through fibrinogen bound to αIIbβ3.48,76

Platelet-leukocyte interactions promoting atherogenesis. (A) Platelet-mediated leukocyte recruitment and adhesion. Leukocyte recruitment to endothelial-bound platelets occurs in a multistep coordinated process. Initial tethering of leukocytes is mediated by the interaction of P-selectin expressed on the platelet surface with its cognate receptor leukocyte PSGL-1. Ligation of PSGL-1 promotes activation of leukocyte β2 integrins (Mac-1 and LFA-1), necessary for stable leukocyte adhesion. Note that Mac-1 can engage several platelet ligands, including GPIbα, ICAM-2, and JAM-3, as well as fibrinogen bound to αIIbβ3. Release of the chemokine RANTES at sites of atherogenesis leads to enhanced monocyte adhesion. Monocytes subsequently migrate through the endothelium and differentiate into macrophages, which engulf lipids and transform into foam cells. (B) Platelet-leukocyte cross-talk. Bioactive mediators derived from α-granules of activated platelets, including PF4, synergize with RANTES to enhance leukocyte adhesion and monocyte differentiation. RANTES, acting in concert with P-selectin, results in MCP-1 and IL-8 secretion by monocytes. CTAP-III is another chemokine released from the platelet α-granules, which is subsequently converted into active NAP-2 by the neutrophil membrane associated serine-protease cathepsin G. NAP-2 enhances leukocyte adhesion and neutrophil transendothelial migration. Up-regulation of COX-2 in monocytes via pathways involving IL-Iβ and P-selectin results in increased production of proinflammatory mediators, including PAF, leukotrienes, and TxA2 via transcellular eicosanoid metabolism. P-sel indicates P-selectin; AA, arachidonic acid.
Platelet-leukocyte interactions promoting atherogenesis. (A) Platelet-mediated leukocyte recruitment and adhesion. Leukocyte recruitment to endothelial-bound platelets occurs in a multistep coordinated process. Initial tethering of leukocytes is mediated by the interaction of P-selectin expressed on the platelet surface with its cognate receptor leukocyte PSGL-1. Ligation of PSGL-1 promotes activation of leukocyte β2 integrins (Mac-1 and LFA-1), necessary for stable leukocyte adhesion. Note that Mac-1 can engage several platelet ligands, including GPIbα, ICAM-2, and JAM-3, as well as fibrinogen bound to αIIbβ3. Release of the chemokine RANTES at sites of atherogenesis leads to enhanced monocyte adhesion. Monocytes subsequently migrate through the endothelium and differentiate into macrophages, which engulf lipids and transform into foam cells. (B) Platelet-leukocyte cross-talk. Bioactive mediators derived from α-granules of activated platelets, including PF4, synergize with RANTES to enhance leukocyte adhesion and monocyte differentiation. RANTES, acting in concert with P-selectin, results in MCP-1 and IL-8 secretion by monocytes. CTAP-III is another chemokine released from the platelet α-granules, which is subsequently converted into active NAP-2 by the neutrophil membrane associated serine-protease cathepsin G. NAP-2 enhances leukocyte adhesion and neutrophil transendothelial migration. Up-regulation of COX-2 in monocytes via pathways involving IL-Iβ and P-selectin results in increased production of proinflammatory mediators, including PAF, leukotrienes, and TxA2 via transcellular eicosanoid metabolism. P-sel indicates P-selectin; AA, arachidonic acid.
Platelet activation with consequent degranulation results in the liberation of numerous chemokines stored in platelet α-granules. These chemokines can either be released into the circulation or displayed on the platelet surface, where they exert numerous biological activities instrumental in atherosclerosis.77,78
CXC chemokines NAP-2 and PF4 (Figure 4)
The two most abundant CXC chemokines stored in platelet α granules are CXCL7 and platelet factor 4 (PF4). CXCL7 has several molecular variants, including the inactive precursor, connective tissue-activating peptide III (CTAP-III), which is enzymatically converted into active neutrophil activating protein-2 (NAP-2) through proteolysis by the neutrophil membrane-associated serine-protease cathepsin G.79 NAP-2 has been demonstrated to induce neutrophil and monocyte adhesion in vitro and neutrophil transendothelial migration.78,80 The chemotactic properties of PF4 on its own are controversial because it does not exert classical chemokine functions at submicromolar concentrations.78,81 However, PF4 acting in concert with other chemokines such as the platelet-derived CC chemokine regulated upon activation normal T-cell expressed and secreted (RANTES) may enhance leukocyte adhesion and promote monocyte differentiation and atherosclerosis. These PF4-RANTES heterodimers have been proposed to represent potential therapeutic targets in the treatment of atherosclerosis.82
CC chemokine RANTES (Figure 4)
The proinflammatory CC chemokine RANTES is released from activated platelets and deposited on activated endothelium, where it is able to promote adhesion of monocytes.83 A prerequisite for the in vivo activity of RANTES function is for it to bind to glycosaminoglycans and to form higher-order oligomers.84 Further, through its interactions with other modulators, including MCP-4, PF4, and P-selectin, RANTES has been implicated in inflammation, atherogenesis, and vascular remodeling after injury. Interestingly, P-selectin engagement with PSGL-1 on monocytes is insufficient for the induction of MCP-1, requiring the cooperation of platelet-derived RANTES for chemokine synthesis by monocytes.85 Further, monocytes costimulated by RANTES and P-selectin secrete a different cytokine profile, including MCP-1 and IL-8.85 Platelet activation results in the release of microparticles that are able to facilitate the deposition of RANTES on the inflamed and atherosclerotic endothelium, thus promoting monocyte adhesion.86
Proinflammatory lipids (Figure 4)
In addition to being storage houses for cytokines and chemokines, platelets also generate lipid-derived inflammatory mediators. The adhesion of platelets to leukocytes and the endothelium results in transcellular metabolism of eicosanoids, leading to the local synthesis of proinflammatory lipids such as PAF, leukotrienes, and TxA2.87 The imbalance of inflammatory mediators is further exacerbated by monocyte up-regulation of COX-2 expression via interactions involving P-selectin derived from activated platelets and IL-Iβ. This up-regulation of COX-2 enables further synthesis of proinflammatory eicosanoids.88 PAF is a potent platelet agonist and inflammatory mediator and has also been shown to regulate firm neutrophil adhesion on the surface of immobilized spread platelets.44
Atherothrombosis and disturbed blood flow
An important determinant influencing platelet-endothelial and platelet-leukocyte interactions at atherosclerotic-prone sites is the local blood flow conditions. It is well established that atherosclerotic lesions develop at arterial branch points, which are typically areas of low shear and disturbed flow. Altered flow has a significant effect on the adhesive properties of platelets and leukocytes, as well on the morphology and function of endothelial cells. Rheological factors also contribute to the deposition of lipids and other modulators of atherosclerosis.
Throughout much of the vasculature, blood flow is laminar, with adjacent fluid layers traveling parallel to each other but at different velocities because of the fluid drag exerted by the vessel wall. This phenomenon leads to the generation of shear forces between adjacent fluid planes. These flow profiles apply to straight, healthy vessels—at arterial branch points, curvatures, and areas of stenosis, flow profiles become significantly more complex. These alterations in flow produce shear gradients, turbulence, flow separation, and eddy formation, each of which can affect the atherogenic process. Progression of the atherosclerotic lesion exacerbates flow disturbances, thus inducing a potential hazardous cycle of shear-dependent acceleration of atherosclerosis.89
Shear effects on the endothelium
Endothelial cells in arteries are constantly exposed to dynamic alterations in shear due to the pulsatile nature of blood flow.90 However, the rate of fluctuation in blood flow appears to be the major parameter altering endothelial cell function. Endothelial cells respond to flow changes through a variety of mechanotransduction mechanisms, including mechanically gated ion channels and G proteins, force-dependent alterations in membrane fluidity, nuclear deformation, cytoskeletal-dependent junctional signaling, and via primary cilia and the glycocalyx.91 These mechanosensory signaling mechanisms are exquisitely sensitive to changes in wall shear stress, leading to alterations in cell morphology, gene-expression profiles, and increased adhesiveness.92
Atheromatous lesions in permissive flow environments for leukocyte adhesion
The mechanisms by which disturbed blood flow influences leukocyte adhesion mechanisms remain ill defined. In contrast to platelets, leukocytes adhere preferentially at low shear regions in the vasculature, typically at wall shear rates < 1000/s. Whereas a minimum threshold shear is required for leukocyte adhesion,93 there is typically an inverse correlation between wall shear and the efficiency of leukocyte recruitment, with optimal adhesion around 150/s.93 The site density of P- and E-selectin molecules also plays a major role in regulating the efficiency of leukocyte adhesion; therefore, the prevailing flow conditions and the degree of P-selectin expression on the surface of activated endothelial cells and endothelial-bound platelets are likely to be major determinants regulating leukocyte recruitment to atherogenesis-prone sites.94 Experimental models of disturbed blood flow lead to enhanced leukocyte accumulation in low shear recirculation zones,95 supporting the concept that flow disturbances may facilitate leukocyte recruitment to atherosclerotic sites.
Impact of high shear and disturbed blood flow on platelet adhesive function
The importance of shear forces in regulating platelet adhesive function has long been recognized. At normal wall shear rates (20-2000/s), shear does not stimulate the activation of platelets in suspension. However at high stress rates (> 5000/s) as occurs in stenosed atherosclerotic arteries, shear can directly induce platelet activation.5 Based on in vitro findings, shear-induced platelet activation is critically dependent upon the sequential binding of VWF to GPIbα and integrin αIIbβ3, as well as on the release of ADP.96 Shear-induced platelet activation is insensitive to COX inhibitors, a finding that may partly explain the relative weak antithrombotic effect of aspirin.97 In a complex shear environment characterized by rapid phases of shear acceleration and deceleration, platelet aggregation can occur independently of ADP and TxA2,98 raising the possibility that shear gradients can promote platelet deposition and initial thrombus growth even in the presence of aspirin and a P2Y12 inhibitor.
Conclusion
It has long been recognized that arterial thrombosis primarily represents an exaggerated physiological hemostatic response at sites of atherosclerotic plaque disruption. A similar concept can be developed for the role of platelets in the initiation and propagation of atherosclerosis. Platelets normally interact with endothelial cells to preserve their physiological function and to enhance leukocyte recruitment to sites of inflammation. An exaggeration of these physiological responses at atherosclerosis-prone sites appears to play an important role in the pathogenesis of this disease. This improved understanding of the role of platelets in the development of atherothrombosis and elucidation of the key pathways involved should stimulate further investigation into the possibility of targeting both the proinflammatory and prothrombotic function of platelets to reduce the clinical complications of this major disease.
Acknowledgments
We thank Dr Simone Schoenwaelder for assistance with the preparation of the figures. Z.S.K. is an National Health and Medical Research Council (NHMRC) and National Heart Foundation postgraduate scholarship holder. S.P.J. is a NHMRC Australia Fellow.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Shaun P. Jackson, Australian Centre for Blood Diseases, 6th Level Burnet Bldg, AMREP, 89 Commercial Rd, Melbourne, Victoria, Australia 3004; Phone: 61-3-9903-0131; Fax: 61-3-9903-0228; e-mail: shaun.jackson@monash.edu.
