
Abstract
Aplastic anemia remains a diagnosis of exclusion. Our ability to reliably diagnose, and therefore exclude, a variety of inherited or acquired diseases with similar phenotypes has improved markedly. An efficient diagnostic plan is important because time from diagnosis to treatment is related to outcome regardless of the therapeutic option chosen. HSCT remains the mainstay of therapy for those with matched sibling donors, and results have improved even further in recent years. For those without a sibling donor, the high response and overall survival rates of combined immunosuppressive therapy (IST) have proven robust. Nonetheless, incomplete response, relapse, and progression to myelodysplasia/leukemia have more clearly emerged as significant long-term issues. Improvements in outcome of alternative donor transplantation and the use of established and novel immunosuppressive agents provide multiple alternatives for treating refractory or relapsed patients. Best practices in this regard are not yet clearly established and may vary by a variety of demographic and treatment-specific factors. Regardless of the type of therapeutic approach, patients require ongoing monitoring for occurrence of disease and/or therapy-related side effects.
Introduction
Aplastic anemia remains a disorder confined by conventions that specify a combination of low peripheral blood counts with specific appearances of the BM itself. The most common conventions, modified by severity criteria, are shown in Table 1. Whereas it has always been thought that BM failure meeting these criteria could arise as a consequence of diverse pathophysiologic mechanisms, the details surrounding some of these mechanisms—both their commonalities and their singularities—are becoming better understood. The intention of this review is to use specific examples of new observations, ways in which existing information is being used in new ways, and some current fields of investigation to illustrate areas of progress or controversy that either are influencing or may come to influence the diagnosis and management of aplastic anemia patients in the near term.
Definition of aplastic anemia with severity criteria*
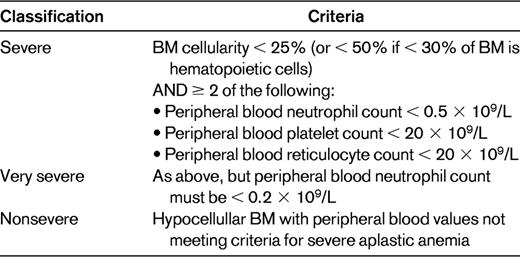
Adapted with permission from: Davies JK, Guinan EC.50
Diagnosis
Despite the precision of its diagnostic criteria, aplastic anemia has always been a diagnosis of exclusion. No single test allows us to reliably diagnose idiopathic aplastic anemia, but the field has advanced considerably in terms of awareness of and diagnosis of other disorders resulting in a similar or indistinguishable hematologic phenotype.1–4 Consequently, the diagnostic evaluation has become increasingly detail driven in its attempt to exclude a list of potential alternative etiologies of BM failure. Some of these investigations are quite novel, whereas others are variations on old themes illuminated by new detail. As practitioners, we need to remain alert to emerging changes in diagnostic tools and practices, and to that end, examples of several such diagnostic practice changes are highlighted below.
Whereas the reasons for obtaining a complete blood count may be very diverse, the differential diagnosis of aplastic anemia usually arises from the observation of pancytopenia (Table 2). Although many sophisticated tests are now available, it remains essential to obtain a thorough history and perform a detailed examination. One goal of history taking is to elicit evidence of any drug or toxin exposures that have been associated with BM aplasia. A quick perusal of any online drug database discloses the ubiquity of BM suppression and, albeit less frequently, BM failure as potential adverse events seen after the administration of drugs from virtually every class. The recognition of these associations reflects the circumstances and extent of usage of/exposure to any particular drug or toxin. Therefore, potential exposures should be explored for plausibility in up-to-date databases. Unfortunately, no tests permit ascertainment of causal relationships between any specific exposure and subsequent BM failure. From the perspective of the individual patient, any associations therefore remain presumptive, and the utility is simply in removing any ongoing exposure. From a population perspective, however, aggregated information may inform post-marketing decisions governing drug labeling. Moreover, Web-based communication platforms make the resulting information readily available for use in treatment decisions. Accordingly, physicians should be aware of and use national programs to report such associations. In the United States, one such resource is the Food and Drug Administration's MedWatch program (http://www.fda.gov/Safety/MedWatch/HowToReport/ucm085568.htm).
Differential diagnosis of peripheral pancytopenia
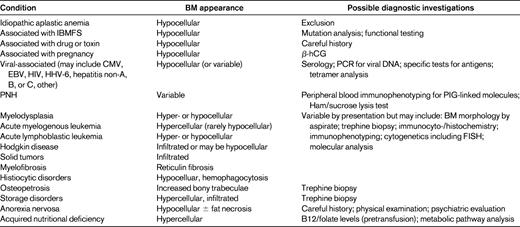
hCG indicates human chorionic gonadotrophin; HHV-6, human herpes virus-6; PIG, XXXX.
Adapted with permission from Davies and Guinan.50
The importance of a detailed physical examination has also not declined. However, there are substantive limitations of the examination in firmly excluding alternative diagnoses previously felt to have pathognomonic findings.5,6 Fortunately, highly specific diagnostic tests have emerged for multiple disorders that can present as aplastic anemia. Genetic testing has proven fruitful in several regards, particularly in providing genetic (mutation identification) tests for inherited BM failure syndromes (IBMFSs). These tests and their application will be discussed in more detail by Dr Niemeyer (see pages 84-89).7 The generalizable message in regard to using these tests to exclude diagnoses other than idiopathic aplastic anemia is that they provide incomplete information. We do not yet know all of the inherited genetic variants that can result in a clinical BM failure phenotype, or in a specific IBMFS phenotype.8,9 Classical mutations can be found in individuals without physical findings of an IBMFS and the same mutation can be associated with very diverse clinical presentations.5,9 Whereas better diagnostic algorithms using phenotypic, clinical laboratory, and genetic data are evolving rapidly and new genetic variants continue to be discovered, current testing does result in improved but not absolute exclusion of a specific IBMFS.7 A different example of the expanded tool kit provided by genetic testing is a recent report in which an infant presented with pancytopenia and BM failure without megaloblastic changes (albeit with some dysplasia) and normal B12 and folate levels. Eventual evaluation of metabolic status led to the finding of a novel transcobalamin mutation resulting in B12-responsive BM failure.9
Routine cytogenetic testing has further revealed that approximately 10% of patients with apparent aplastic anemia by all other criteria may have clonal chromosomal abnormalities. The complex relationship of clonality and aplasia is presented in great detail by Dr Maciejewski (see pages 90-95).11 Additional data based on cell-surface and intracellular markers such as p53, Hgb F and telomere length are also being evaluated for their utility in the diagnostic quagmire of differentiating hypoplastic myelodysplasia and idiopathic aplastic anemia.12,13
Innovations in established diagnostic algorithms have become commonplace. One paradigm is investigation of paroxysmal nocturnal hemoglobinuria (PNH) as a cause of aplasia. Testing for PNH has evolved significantly from function-based biochemical assays such as the sucrose hemolysis and Ham tests to flow cytometric analysis.14 Somewhat confusingly from a diagnostic point of view, both healthy individuals and those with aplastic anemia can have clones of cells with a PNH phenotype, and these clones can wax and wane in absolute and relative frequency.14 Therefore, accurate biomarker identification in conjunction with accurate quantitation has proven requisite. Both goals have been met by flow cytometric detection and quantification of PNH clones by use of the glycophosphatidylinositol-anchor binding, fluorescently labeled inactive toxin aerolysin, which is more sensitive than antibody binding to CD59, another glycophosphatidylinositol-anchor-binding cell-surface molecule.14
Mutation analysis is a highly specific but incomplete mechanism for establishing an alternate IBMFS diagnosis. However, testing for a common functional or structural phenotype can be highly synergistic and facilitative. Just as abnormal sister-chromatid exchange became the diagnostic test for virtually every Fanconi anemia patient, determination of telomere length has become a valuable adjunct to diagnosis of dyskeratosis congenita in particular, but to other IBMFSs as well.15,16 Using several different techniques, nomograms of telomere length by age and by cell of origin are becoming sufficiently refined to provide significant diagnostic assistance. Differences in cell-cycle markers may also prove useful.17 As additional biological correlates of IBMFS molecular findings become better understood, such “functional” screening should become both more robust and accessible.
Management
Whereas there has been no significant shift in the management strategy for aplastic anemia over the last several years, there are some emergent data that can support and direct the practitioner in managing such patients.
Supportive care
Medical care continues to depend upon meticulous attention to issues of infectious and hemorrhagic diatheses, expectant management of regimen-related toxicities, and provision of information and psychological support.1–4 Potentially useful data drawn from related patient populations or those with similar issues emerge routinely. For example, increased scrutiny of platelet transfusion triggers in diverse populations, few of whom have aplasia with its attendant protracted platelet production failure, has been undertaken but should be interpreted cautiously for this population.18 Similarly, the approach to potential infection in neutropenic, febrile patients is frequently updated and provides important algorithms, but its applicability is limited by the persistent pancytopenia of aplastic anemia patients compared with other populations. However, the spectrum of infections in aplastic patients specifically has been reviewed recently and treatment recommendations have been provided.19
Emerging data on the use of iron chelation in patients with aplastic anemia
Iron-related mortality, especially related to hepatic and cardiac dysfunction, has not surprisingly emerged as an issue in aggregated BM failure cohorts, including some individuals with aplasia.20,21 Daily chelation with oral deferasirox has been studied prospectively in a large number of aplastic anemia patients with iron overload. Treatment was well tolerated and effective in decreasing serum ferritin and transaminases.21 Expectant, cautious management was urged in regard to renal impairment, especially if there was concomitant use of renal toxic immunosuppressive drugs.21 In addition to producing desired improvements in organ function,20,21 in a few cases, chelation with either deferasirox or deferoxamine has also intriguingly been associated with significant hematologic improvement.22,23
Matched family member HSCT
Mainstays for treatment for aplastic anemia remain HSCT, the only curative therapy to date, and immunosuppressive therapy (IST). HSCT continues to be the recommended first-line therapy for individuals with severe or very severe aplastic anemia who have a matched sibling donor.1–4 The upper limit of age for this recommendation has been 40 years, although there is increasing variation in this regard, especially with use of less-aggressive conditioning regimens. Results of matched sibling HSCT have improved over time. A large, recent retrospective review found a significantly inferior overall survival rate of 73% (n = 614) in those patients receiving transplantations between 1991 and 1996 compared with 80% (n = 550) in those receiving transplantations between 1997 and 2002.24 Survival of children in the latter cohort was even higher at 91%. Indeed, younger age, year of transplantation, and decreased interval from diagnosis to transplantation all contributed to improved outcome in this European registry report. Conditioning regimens for matched sibling HSCT have historically been limited in their reliance on radiation, a trend that has become more pronounced. For example, 24% compared with 8% of matched sibling HSCT incorporated radiation into the conditioning regimen in the above 2 time periods, respectively, and irradiation was inversely correlated with survival.24 The mainstay of conditioning has remained cyclophosphamide with or without additional agents.
Immunosuppressive therapy
For individuals lacking a matched sibling donor or above the age at which sibling HSCT is felt to represent the best opportunity for good outcome, IST is indicated.1–4 The multiagent regimen of antithymocyte globulin (ATG) and cyclosporine (generally accompanied by a brief course of corticosteroids) has proven very robust.25 Response to IST generally ranges from 50%-80% and, in contrast to HSCT, the response rate of patients to IST has not changed in recent years.1,2,24,26–28 IST regimens are somewhat variable, particularly with respect to source and administration schedules of ATG as detailed in the cited reports and reviews. Most studies have used horse ATG, although for various reasons more practical than theoretical, rabbit ATG is currently also in use. No substantial data as to the relative efficacy of the preparations have yet emerged. Modifications to the conventional IST regimen, including addition of danazol,29 mycophenolate mofetil,30 sirolimus,27 or hematopoietic growth factors,31 have not significantly improved response or decreased relapse rates. Such agents currently have no place in primary therapy, although a few studies suggest that the addition of danazol29 or growth factors29,31 has altered relapse rates. Very little information is available about the substitution of tacrolimus for cyclosporine.32 Alternative immunosuppressive regimens, such as cyclophosphamide33,34 or alemtuzumab with or without cyclosporine,35 also show promise. Most IST modifications have been studied in very limited cohorts and have not yet been subjected to prospective, randomized comparisons with the standard of care.
With current improvements in alternative donor HSCT, the desire to predict response to IST has grown. Several recent reports have suggested that younger age overall is associated with greater likelihood of response.1,24,36 Among affected children only, younger age has inconsistently been associated with greater response rate.1,2 Interval from diagnosis to treatment, gender, absolute neutrophil count (ie, very severe aplastic anemia vs severe aplastic anemia), and reticulocyte and lymphocyte counts have also, but more variably, been correlated with response.1,2,24,28 Telomere length has not been shown to be correlated with response.13
Scrutiny of the definition of response chosen, which varies between investigators and programs, is important in interpreting the results of IST studies; the widespread adoption of consensus criteria should facilitate comparison of outcomes. In addition to varied degree of improvement, IST responders can follow highly variable clinical paths. Some are stable after discontinuation of treatment, whereas others (15%-25%) have significant cyclosporine dependence.26 Slower weaning off of cyclosporine (over approximately 1 year) has been associated with decreased relapse.26 Some patients remain stable or slowly improve their hematologic status, whereas others may relapse (apparently) spontaneously or with “provocations” such as pregnancy, and still others go on to develop persistent clonal cytogenetic findings and myelodysplasia or acute myelocytic leukemia. This latter topic is reviewed and referenced in detail elsewhere in this book, and is an important component of both managing and triaging treatment in aplastic anemia patients. For patients who are refractory to IST or who relapse after successful IST, treatment with an additional course of ATG-based IST is possible.3,4,29,37,38 Response rates in recent reports range from 11%-65%,29,37,38 with rates in previous responders generally more favorable than in initially refractory patients. More experimental modalities, including alternative IST (eg, rituximab39 or cyclophosphamide34 ) may also be considered. In addition to an understanding of the diverse courses of patients after IST, which range from uninterrupted hematologic stability to either relapse or evolution of myelodysplasia/leukemia (both at very unpredictable times after treatment), these choices should be determined by factors including patient age, performance status, transfusion requirement, comorbid conditions, and availability of alternative donors for HSCT. Intriguingly, shorter age-adjusted telomere length has been shown recently to be associated with likelihood of relapse, clonal evolution, and overall mortality.13
Alternative donor HSCT
The above choice is heavily influenced by the recent significant improvement in the outcome of alternative donor transplantation, using either matched or mismatched unrelated donors (URDs) or mismatched related donors. In part because of the underlying historical tendency of both alternative donor and aplastic anemia patients to have increased graft failure/rejection, alternative donor transplantations for aplasia were initially carried out with aggressive, immunosuppressive, and myeloablative regimens, markedly different from those used for matched sibling HSCT. Moreover, the aplastic anemia patients who first came to URD were often heavily treated patients who had failed numerous therapies, had repeated episodes of febrile neutropenia or infection, and had received extensive transfusion support. In a multivariate analysis of the European registry data, in which actuarial survival after alternative donor HSCT improved from 38% to 65% in the periods 1991-1996 and 1997-2002, respectively, only year of transplantation was associated with increased survival.24 It is likely that progressive changes in dimensions such as improved performance status, decreased prior transfusion, decreased interval from diagnosis to transplantation, improved supportive care, better donor-recipient matching, and use of less-intensive (particularly low-dose radiation or radiation-free) regimens contributed to this association and to the improved results in other recent studies.24,38,40–44 Five-year survival ranges from approximately 35%-85% in these reports, depending upon approach, match, and recipient age. More reliable data on unrelated umbilical cord blood HSCT for aplastic anemia have also started to emerge, although this approach remains used largely in pediatric patients.45,46
Overall, younger, better-matched patients have superior outcomes after alternative donor HSCT.24,38,40–44 However, degree of match or choice of alternative donor has not significantly affected various outcome measures in some studies.42 Attention has recently turned to additional pretransplantation characteristics that might influence or predict outcomes. One example would be consideration of the effects of iron overload at time of HSCT. Because patients with matched sibling donors most often move rapidly to HSCT, these studies are largely referable to alternative donor HSCT. Once again, inferences are drawn from studies in which patients with various diagnoses are aggregated rather than studies in which the diagnosis is confined to those with aplastic anemia. Nonetheless, the majority of aplastic patients in these studies tended to have high ferritin levels or high aggregate “iron scores” at a level the reports associated with increased 100-day mortality, acute GVHD occurrence, bacteremia/infection, and decreased overall survival.47,48 However, the associations between iron overload and adverse outcomes observed may be skewed by the prevailing myelodysplasia diagnosis in the study cohorts, and this and other confounding factors may contribute to these observed outcomes. There is as yet no data in aplastic anemia that directly address the value of chelation-related decreases in iron compared with the natural presentation to HSCT with less iron-loaded status. Whereas it is likely fair to take the issue of iron-related toxicity as another incentive among many to proceeding with treatment quite expeditiously, overall, the consideration of the costs, benefits, and potential toxicities of iron chelation must continue to be considered largely on a patient-by-patient basis.
Despite increased data on the outcome of patients treated in diverse ways, there is as yet no absolute algorithm in regard to treatment for patients relapsed after or refractory to IST. A recent prospective, multicenter study in Japan evaluated the outcome of pediatric patients with either severe or very severe aplastic anemia who failed to respond to IST at 6 months. The patients underwent HSCT if they had a serologically matched URD, an HLA-one antigen–mismatched family donor, or an HLA-matched or HLA-one antigen–mismatched umbilical cord blood donor at the time of evaluation or they received a second IST course identical to the first.38 The failure-free survival (ie, survival with hematologic response) at 5 years was 83.9% in those receiving HSCT, significantly better than 9.5% in those given a second round of IST. With the caveat that the epidemiology of aplastic anemia and HSCT in Japan has been somewhat different from that in other geographic areas, these are nevertheless important and provocative data. Results that can be inferred from prior, retrospective reports are somewhat less divergent than this, albeit not in direct contrast. Additional analyses based on patients treated over the last decade will help to inform better decision making in regard to secondary treatment.
Long-term toxicity
The long-term complications of HSCT (somewhat independent of underlying disease) are increasingly well appreciated,49 and long term survivors of IST1,29 are also at risk for a multiplicity of complications. Some of these complications become obvious only with prolonged followup. Moreover, these data are imperfect because both the characteristics of specific patient cohorts and regimens influence outcome. One might expect that as patients with different diseases, particularly IBMFS, are removed from the “aplastic anemia” cohort and as regimens alter (certainly toward less radiation and potentially to alternative IST), the challenges faced by survivors will also change.
Summary
Incremental gains have been made in both the diagnosis and management of aplastic anemia. The issues of predicting treatment response and clinical course remain unresolved, although some intriguing data have started to emerge. Greater understanding of regimen-related toxicities, either acute or delayed and potentially chronic, provides an impetus for the improvement of therapeutic strategies.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Very few of the drugs used to treat aplastic anemia are approved for that purpose (eg, cyclosporine, cyclophosphamide).
Correspondence
Eva Guinan, MD, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA 02215; Phone: 617-632-4932; Fax: 617-632-3770; eva_guinan@dfci.harvard.edu.
