
Abstract
Hypoplastic BM disorders in children and adolescents comprise a broad spectrum of disorders. Acquired severe aplastic anemia (SAA), refractory cytopenia of childhood (RCC), a subtype of myelodysplastic syndrome (MDS), and inherited BM failure (IBMF) disorders are the main and most difficult hematological differential diagnoses. Whereas IBMF disorders can often be diagnosed by their clinical features and/or underlying genetic aberrations, the morphological distinction between SAA and hypocellular RCC has been controversial. The histopathological pattern of RCC consists of islands of immature erythroid precursors accompanied by sparsely distributed granulocytic cells. Megakaryocytes are significantly decreased or absent and, rarely, micromegakaryocytes are detected on immunohistochemistry. Because fatty tissue between areas of hematopoiesis can mimic SAA, 2 biopsies are recommended to facilitate the detection of representative BM spaces. Recent data indicate that the response to immunosuppressive therapy is inferior in RCC compared with SAA. Furthermore, approaches to allogeneic hematopoietic transplantation differ. Controlled prospective clinical studies in patients with hypoplastic BM failure disorders will require comprehensive guidelines for diagnosing SAA, RCC, and the different IBMF disorders.
Introduction
The appropriate classification of hypoplastic BM disorders in young patients continues to be a great challenge. Accurate diagnosis is the prerequisite for medical and genetic counseling, surveillance, and therapy options for these children and adolescents. Because most patients with BM failure will require allogeneic hematopoietic stem cell transplantation (HSCT), the timing of the procedure, choice of donor, preparative regimen, and estimation of complications will greatly differ among the various disease entities.
Hypoplastic BM disorders in children and adolescents
Pancytopenia with severe decrease of BM cellularity in children may be caused by a broad variety of underlying disorders (Table 1); of these, acquired severe aplastic anemia (SAA), refractory cytopenia of childhood (RCC), and inherited BM failure (IBMF) disorders are 3 of the most common hematopoietic diagnoses. The clinical and histopathological distinction between these 3 groups of disorders is a well-known challenge and has important therapeutic implications. Whereas SAA is considered an autoimmune-driven BM failure disorder for which most children are successfully treated with immunosuppressive therapy (IST),1,2 RCC is classified as a subtype of myelodysplastic syndrome (MDS) generally thought to be cured by HSCT only.3 IBMF is likely to be underdiagnosed. With the recent discoveries of underlying genetic alterations, these disorders are being diagnosed with increasing frequency in patients without phenotypic abnormalities.4

Small populations of cells deficient in glycophosphatidylinositol-anchored proteins may be noted in the absence of clinical and laboratory evidence of hemolysis in all 3 groups of BM failure disorders. They are most common in SAA, for which 40%-50% of patients carry these clones characteristic of paroxysmal nocturnal hemoglobinuria (PNH) at the time of diagnosis.5 Response rate to IST is similar in SAA patients with or without PHN clones. In contrast to subclinical disease, overt PNH with thrombosis and hemolysis in the setting of BM failure is a rare finding in childhood.6
Pancytopenia with a moderately or severely hypocellular BM may also herald acute leukemia. B-cell precursor acute lymphoblastic leukemia is preceded by a transient, remitting aplasia in ∼ 2% of childhood acute lymphoblastic leukemia.7 Hematopoietic cells in the aplastic phase generally have a normal karyotype, but may carry a large number of secondary genetic changes characteristic of overt leukemia diagnosed up to 9 months later.8 Rarely, familial and acquired hemophagocytic lymphohistiocytosis9 or autoimmune lymphoproliferative disorder10 may give rise to hypocellular BM.
These genuine BM disorders can be mimicked by a large variety of nonhematological diseases such as viral infections, nutritional deficiencies, and metabolic disorders (Table 1). A careful medical history and presence of physical signs such as hepatosplenomegaly or lymphadenopathy may suggest an infectious origin. Interestingly, persisting or transient BM failure after varicella infection may be accompanied by multiple autoantibodies.11
Classification of pediatric MDS
The need for a pediatric approach to the diagnosis and management of MDS in children and adolescents has emerged over the past 2 decades.12 The pediatric modification12 of the first World Health Organization (WHO) classification13 subdivided MDS into RCC (peripheral blood [PB] blasts < 2% and BM blasts < 5%), refractory anemia with excess blasts (RAEB; PB blasts 2%-19% and/or BM blasts 5%-19%) and RAEB in transformation (RAEB-T; PB and/or BM blasts 20%-29%).12 This approach allows unambiguous classification of > 95% of patients14 and was incorporated into the second WHO classification in 2008.15
MDS after prior chemotherapy or radiation therapy, after prior acquired aplastic anemia, or in IBMF disorders and familial diseases is generally classified as secondary MDS. All other cases are referred to as primary MDS, although it is reasonable to assume that most of these disorders are secondary to some as-yet-unknown genetic predisposition. These presumed underlying genetic changes may also give rise to subtle phenotypic abnormalities observed in many children with primary MDS.12
Monosomy 7 is the most common cytogenetic abnormality in childhood MDS, being seen in ∼30% of cases.16–18 Trisomy 8 and trisomy 21 are the second most common numerical abnormalities. Constitutional trisomy 21 is usually clinically obvious when present, whereas constitutional trisomy 8 mosaicism may remain unrecognized and should be tested when trisomy 8 is found in the BM.19 The presence of a structurally complex karyotype characterized by ≥ 3 chromosomal aberrations, including at least 1 structural aberration, is the strongest independent prognostic marker predicting poor outcome.18
RAEB in children has similar morphological and immunophenotypical features to that observed in adults. Children with RAEB generally have relatively stable counts for weeks or months. A similar biological behavior is noted in some children considered RAEB-T in the French-American-British (FAB) Cooperative Group classification. Because these children may not benefit from acute myeloid leukemia–type intensive chemotherapy before HSCT, the subdivision of RAEB-T was retained in the pediatric MDS classification.12,15
RCC
RCC is the most common subtype of MDS in children and adolescents, accounting for approximately half of all MDS cases.17,20 It is diagnosed in all age groups and, in contrast to MDS with increased blast count, affects boys and girls with equal frequency.21
Children usually present with symptoms related to pancytopenia, such as anemia, infection, and bleeding tendency. The term “refractory cytopenia” indicates that, in contrast to adults, thrombocytopenia and neutropenia are more frequently observed than anemia in children. In fact, 3/4 of patients present with a platelet count < 150 × 109/L, 1/2 with an absolute neutrophil count at the time of diagnosis of < 1.0 × 109/L and 1/4 of 0.5 × 109/L. Anemia with a hemoglobin concentration of < 10 mg/100 mL is seen in approximately 1/2 of affected children.21 The mean corpuscular volume of red cells and hemoglobin F are usually elevated. In contrast to adult refractory anemia, the majority of children with RCC show a marked decrease of BM cellularity. In an interim analysis of studies, the European Working Group of MDS of Childhood (EWOG-MDS) 1998 and 2006 found that 81% of patients with RCC had a hypocellular biopsy specimen (Figure 1) (EWOG-MDS, unpublished data).

BM cellularity and karyotype in primary RCC. Interim analysis of studies EWOG-MDS 1998 and 2006.
BM cellularity and karyotype in primary RCC. Interim analysis of studies EWOG-MDS 1998 and 2006.
Because a variety of nonhematological disorders can induce secondary myelodysplasia, BM aspiration cytology constitutes only one aspect of the RCC diagnosis. In particular, the need for the differentiation from other hypocellular BM disorders requires the evaluation of a trephine biopsy. In the absence of a cytogenetic marker, the clinical course will have to be carefully evaluated and 2 BM examinations with biopsies at least 2 weeks apart are recommended before a diagnosis of RCC can be established.
The presence of a cytogenetic abnormality can be important in confirming the diagnosis of MDS. Because of the difficulty in establishing the diagnosis of childhood RCC, it is likely that in retrospective studies, RCC patients with chromosomal changes were preferentially reported.17,21 In the prospective multicenter study EWOG-MDS 98, standard cytogenetics in BM cells of patients with RCC and normal or increased BM cellularity showed a normal karyotype, monosomy 7, or other aberrations in 61%, 19%, and 12% of cases, respectively (Figure 1; EWOG-MDS, unpublished data). In hypocellular RCC, the incidence of monosomy 7 and other aberrations decreased to 20% of successful analyses (Figure 1). There was no difference in morphology between cases with or without monosomy 7 or other aberrations. Loss of the long arm of chromosome 5 (5q−), the most frequent chromosomal aberration in adults with refractory anemia, is exceedingly rare in childhood. Studies are under way to evaluate whether newer techniques such as single-nucleotide polymorphism array-based karyotyping can be important in distinguishing RCC and SAA.
Spontaneous disappearance of cytopenia with monosomy 7 or deletions of the long arm of chromosome 7, del (7q), has been reported in some infants, but remains a rare event.22,23 Although chromosomal changes are not thought to be initiating events, they are probably involved in disease progression, and karyotypic evolution is generally accompanied by progression to more advanced forms of MDS in each case. Patients with monosomy 7 have a significantly higher probability of progression to advanced MDS than do patients with other chromosomal abnormalities or normal karyotype.21 In a retrospective series of 67 patients with RCC, the estimated median time to progression for children with monosomy 7 was 1.9 years and the cumulative incidence of progression was 80% at 6 years from diagnosis. In contrast to monosomy 7, patients with trisomy 8 or normal karyotype may experience a long, stable course of their disease.
Morphological distinction between hypoplastic RCC and acquired aplastic anemia
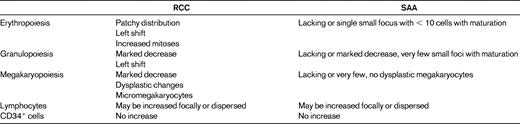
Hypoplastic RCC shows a pathohistological pattern of the BM that is comparable to hyper- or normocellular RCC (Figure 2). In both, the detection of erythroids with impaired maturation is essential and is an important marker for the diagnosis of RCC. In hypoplastic RCC, one or possibly several clusters of at least 10 erythroid precursors with increased numbers of proerythroblasts and increased numbers of mitoses are distributed in a patchy pattern in an otherwise adipocytic BM.15 Granulopoiesis is always decreased and may be left shifted as well. Megakaryocytes are markedly decreased or absent. Immunohistochemistry is indispensable for the detection of micromegakaryocytes, which can be easily be missed on hematoxylin and eosin stains of the BM trephine or aspirate. In contrast to MDS in adults, RCC shows no increase in reticulin fibers. Lymphocytes, plasma cells, and mast cells may be increased. Clusters of blast cells are not seen in RCC, and myeloperoxidase-, lysozyme-, CD34-, and/or CD117-positive myeloid blast cells are not increased.

Schematic view of the histopathological pattern in RCC with normocellular and hypocellular BM.
Schematic view of the histopathological pattern in RCC with normocellular and hypocellular BM.
Acquired aplastic anemia reveals aplasia of all 3 hematological cell lineages. Lymphocytes, plasma cells, and mast cells may be focally increased or dispersed. Erythroid islands with an increased number of immature erythroblasts, particularly proerythroblasts and micromegakaryocytes, are absent (Table 2). After immunosuppressive therapy, the histological pattern of acquired aplastic anemia can no longer be separated from that observed in RCC.
Histopathologic criteria of RCC and SAA as published in the WHO classification
(Used with permission from Baumann et al.15 )
RCC and inherited BM failure disorder
IBMF disorders with pancytopenia, such as Fanconi anaemia, dyskeratosis congenita, Shwachman Diamond syndrome, amegakaryocytic thrombocytopenia, and pancytopenia with radio-ulnar synostosis, show overlapping morphological features with RCC. There is macrocytosis of red cells on PB smear, elevated hemoglobin F, and undistinguishable histopathological findings on trephine biopsies.15 Therefore, IBMF disorders have to be excluded by careful past medical and family history, meticulous physical examination for skeletal and other organ abnormalities, and the appropriate laboratory/molecular studies before a definite diagnosis of RCC can be made.
For most patients, the suspicion of Fanconi anemia arises only after the development of BM failure, which generally starts in childhood or adolescence.4 Approximately 15% of consecutive German patients presenting for the first time with a histopathological pattern compatible with RCC are later diagnosed as suffering from Fanconi anemia, and in approximately 2/3 of these cases, Fanconi anemia was not the primary suspected diagnosis of the treating physician (EWOG-MDS, unpublished data). Analyses to exclude Fanconi anemia by chromosomal breakage test, G2 cell-cycle arrest, and Western blot or mutational analyses are mandatory for all the children before the diagnosis of RCC can be confirmed.
Similarly to Fanconi anemia, cytopenia can precede the appearance of the characteristic mucocutaneous triad of nail dystrophy, mucosal leucoplakia, and abnormal skin pigmentations or pulmonary symptoms in dyskeratosis congenita.4 In a heterogenous series of patients diagnosed as having acquired aplastic anemia or low-grade MDS, heterozygous mutations in the core component of telomerase, namely the telomerase RNA component (TERC) and the telomerase reverse transcriptase component (TERT), and in the shelterin component TIN2 can be observed in ∼ 5%-10%.24–27 Because telomeres of mononuclear cells in patients with dyskeratosis congenital are invariably short, telomere length is thought to be a suitable screening parameter to select patients for molecular analyses of involved genes.28
Homozygous or compound heterozygous SBDS gene mutations were not noted in screened series of patients with acquired BM failure, indicating that in most patients with Shwachman Diamond syndrome, pancytopenia was not the only presenting clinical feature.29,30 The significance of heterozygosity for SBDS mutations remains to be unraveled.30
In children with congenital amegakaryocytic thrombocytopenia, BM trephine usually has normal absent or low numbers of megakaryocytes but unremarkable erythropoiesis and granulopoiesis at the time of diagnosis.31 As disease progresses, BM hypocellularity with an RCC pattern develops.
The syndrome of proximal radio-ulnar synostosis with BM failure is another rare differential diagnosis of RCC.32 It is conceivable that several children diagnosed with hypoplastic RCC may suffer from an as-yet-undiagnosed IBMF disorder. Because family members may suffer from the same disorder, BM examination with biopsy, cytogenetic examination, and possibly measurement of telomere length is advisable for all potential family HSCT donors irrespective of PB counts.33
Because IBMF disorders cannot be separated from RCC by morphology, progression to MDS in IBMF requires careful definition. Secondary MDS in IBMF can morphologically be diagnosed in the presence of an elevated blast percentage or increasing BM cellularity despite continuing pancytopenia. Cytogenetic abnormalities such as partial or complete deletion of chromosome 7; 21q abnormalities; losses of 11q, 20q, or 5q; and trisomy 8 may also indicate secondary MDS. However, leukemic progression in each of these IBMF disorders is associated with a complex pattern of recurrent chromosomal and genomic abnormalities, some of them specific for the disorders, others commonly found in MDS and secondary acute myeloid leukemia. In Fanconi anemia, a gain of chromosomal segment 3q26 is an adverse risk factor for leukemic progression,34 whereas a sole gain of chromosome 1q may persist for years without development of MDS or acute myeloid leukemia.35 Similarly, persisting or fluctuating clones have been observed in dyskeratosis congenita.4 Patients with Shwachman Diamond syndrome and isochromosome 7q [i(7q)] or del(20q) can have a stable clinical course for years.36 The increased incidence of i(7q) in Shwachman Diamond syndrome may suggest that it is a fairly specific clonal marker in this syndrome and that it may be related to the SDS gene, which is located on the centromeric region of chromosome7 (7p12-7q11). The clinical significance with respect to therapeutic options and clinical benefits of the various cytogenetic and molecular aberrations will need to be carefully evaluated in defined prospective studies in these individual IBMF disorders. Because abnormal clones can potentially regress in any IBMF disorder, close monitoring is advisable.
Treatment options for RCC
HSCT is the treatment of choice for patients with RCC and monosomy 7 or complex karyotypes early in the course of the disease. In view of a low transplantation-related mortality, HSCT can also be recommended for patients with other karyotypes if a suitable donor is available3,20 (Figure 3). For children with hypocellular RCC, a preparative regimen with reduced-intensity conditioning represents an effective transplantation strategy in the absence of chromosomal abnormality.3 A watch and wait strategy with careful observation may be reasonable for patients in the absence of transfusion requirements, severe cytopenia, or infections. Because early BM failure can at least in part be mediated by T-cell immunosuppression of hematopoiesis,37 immunosuppressive therapy can be a successful therapy strategy for some children with RCC.38,39 However, response and failure-free survival after IST is less favorable in RCC compared with SAA.39 In a recent interim report of EWOG-MDS on 91 children given IST as frontline treatment, the response rate at 6 months was 63% and the overall and failure-free survival rates at 5 years were 90% and 40%, respectively.40

Summary and future directions
Improvement of clinical care for patients with BM failure disorders will depend in part on comprehensive diagnostic guidelines with internationally accepted definitions.41 The latter will also allow the prospective enrollment of defined patient populations in registries and therapeutic trials. In the past, the term “aplastic anemia” has been broadly applied to hypocellular BM irrespective of the respective histopathological features. Using the same morphological criteria applied for patients with normal cellularity, the histopathological pattern of hypoplastic RCC of childhood was established. Reference review of diagnostic trephine biopsies before enrollment in the prospective study for aplastic anemia (SAA 84) or MDS (EWOG-MDS 98, 2006) greatly reduced the incidence of early clonal evolution among SAA patients.42 In addition, hypoplastic BM with cytogenetic aberrations was solely classified as RCC (EWOG-MDS, unpublished data). Future research will have to unravel the etiology and biological nature of both SAA and RCC.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Charlotte M. Niemeyer, Department of Pediatric Hematology/Oncology, University Medical Center Freiburg, Mathilden Strasse 1, D-79106 Freiburg, Germany; Phone: +49-761-270-45060; Fax: +49-761-270-45180; e-mail: charlotte.niemeyer@uniklinik-freiburg.de.
