
Abstract
The inborn errors of heme biosynthesis, the porphyrias, are 8 genetically distinct metabolic disorders that can be classified as “acute hepatic,” “hepatic cutaneous,” and “erythropoietic cutaneous” diseases. Recent advances in understanding their pathogenesis and molecular genetic heterogeneity have led to improved diagnosis and treatment. These advances include DNA-based diagnoses for all the porphyrias, new understanding of the pathogenesis of the acute hepatic porphyrias, identification of the iron overload-induced inhibitor of hepatic uroporphyrin decarboxylase activity that causes the most common porphyria, porphyria cutanea tarda, the identification of an X-linked form of erythropoietic protoporphyria due to gain-of-function mutations in erythroid-specific 5-aminolevulinate synthase (ALAS2), and new and experimental treatments for the erythropoietic prophyrias. Knowledge of these advances is relevant for hematologists because they administer the hematin infusions to treat the acute attacks in patients with the acute hepatic porphyrias, perform the chronic phlebotomies to reduce the iron overload and clear the dermatologic lesions in porphyria cutanea tarda, and diagnose and treat the erythropoietic porphyrias, including chronic erythrocyte transfusions, bone marrow or hematopoietic stem cell transplants, and experimental pharmacologic chaperone and stem cell gene therapies for congenital erythropoietic protoporphyria. These developments are reviewed to update hematologists on the latest advances in these diverse disorders.
Introduction
The inborn errors of heme biosynthesis, the porphyrias, are metabolic disorders, each resulting from the deficiency of a specific enzyme in the heme biosynthetic pathway (Figure 1; Table 1).1,2 These enzyme deficiencies are inherited as autosomal dominant, autosomal recessive, or X-linked traits, with the exception of the most common porphyria, porphyria cutanea tarda (PCT), which usually is sporadic. Notably, these are disorders in which environmental, physiologic, and genetic factors interact to alter the normally tight regulation of heme biosynthesis and cause disease.

The human heme biosynthetic pathway. The pathway consists of 8 enzymatic steps: 4 localized in mitochondria and 4 in the cytosol. Only the type III isomers of uroporphyrinogen and coproporphyrinogen are metabolized to heme. Heme is exported from mitochondria for incorporation into cellular hemoproteins and, particularly in liver, exerts feedback regulation on 5-aminolevulinic acid synthase (ALAS1).
The human heme biosynthetic pathway. The pathway consists of 8 enzymatic steps: 4 localized in mitochondria and 4 in the cytosol. Only the type III isomers of uroporphyrinogen and coproporphyrinogen are metabolized to heme. Heme is exported from mitochondria for incorporation into cellular hemoproteins and, particularly in liver, exerts feedback regulation on 5-aminolevulinic acid synthase (ALAS1).
Human heme biosynthetic enzymes and genes
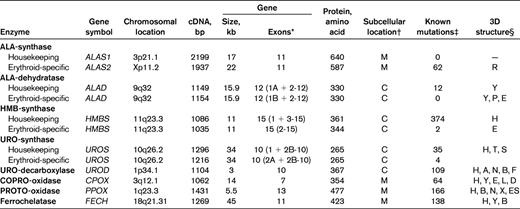
ALAS2: There are 60 loss-of-function mutations in exons 1-11 causing X-linked sideroblastic anemia and 2 gain-of-function mutations causing XLP.
— indicates not applicable.
*Number of exons and those encoding separate housekeeping and erythroid-specific forms indicated in parentheses.
†M indicates mitochondria; and C, cytoplasm.
‡Number of known mutations from the Human Gene Mutation Database (www.hgmd.org) as of May 1, 2012.
§Crystallized from human (H), murine (M), Escherichia coli (E), Bacillus subtilis (B), Rhodobacter capsulatus (R), Pseudomonas aeruginosa (P), Shewanella amazonensis (S), Thermus thermophilus (T), Nicotiana tabacum (N), Aquifex aeolicus (A), Shigella flexneri (F), Leishmania donovani, naafi, and major (L), Desulfovibrio desulfuricans (D), Exiguobacterium sibiricum (ES), Myxococcus xanthus (X), or yeast (Y) purified enzyme; references in Protein Data Bank (www.rcsb.org).
Hematologists often are involved in the diagnosis and/or treatment of these diseases, which as a group can be difficult to diagnose and are considered by many physicians as confusing and complex. Here we present a simplified approach to their classification and focus on the recent advances in understanding their pathogenesis, gene-based diagnoses, identification of their molecular genetic heterogeneity, and currently approved and experimental therapies. We also briefly highlight relevant aspects of heme biosynthesis, including the role of the housekeeping and erythroid-specific 5-aminolevulinate synthase genes (ALAS1 and ALAS2), the mechanism of inhibition of uroporphyrinogen decarboxylase (URO-decarboxylase) activity in sporadic and familial PCT, and the recent recognition that ALAS2 gain-of-function mutations cause X-linked protoporphyria (XLP).
Recent comprehensive reviews of heme biosynthesis and the porphyrias are available.1–4 Informative and up-to-date Web sites are sponsored by the Porphyrias Consoritium of the National Institutes of Health Rare Disease Clinical Research Network (http://rarediseasesnetwork.epi.usf.edu/porphyrias/index.htm), the American Porphyria Foundation (www.porphyriafoundation.com), and the European Porphyria Initiative (www.porphyria-europe.org). An extensive list of unsafe and safe drugs for persons with the acute hepatic porphyrias can be found at the Drug Database for Acute Porphyrias (www.drugs-porphyria.com).
Heme biosynthesis and regulation
Heme is required for a variety of hemoproteins, including hemoglobin, myoglobin, respiratory cytochromes, and the cytochrome P450 enzymes. Hemoglobin synthesis in erythroid precursor cells accounts for ∼ 85% of daily heme synthesis in humans. Hepatocytes account for most of the rest, primarily for synthesis of cytochrome P450 enzymes, which are especially abundant in the liver endoplasmic reticulum, and turn over more rapidly than many other hemoproteins, such as the mitochondrial respiratory cytochromes.
Heme biosynthesis involves 8 enzymatic steps in the conversion of glycine and succinyl-coenzyme A to heme (Figure 1).1,5 The first and last 3 enzymes in the pathway are located in the mitochondrion, whereas the other 4 are in the cytosol. These 8 enzymes are encoded by 9 genes, as the first enzyme in the pathway, ALA-synthase, has 2 genes that encode unique housekeeping and erythroid-specific isozymes, ALAS1 and ALAS2, respectively. In addition, separate erythroid-specific and nonerythroid or “housekeeping” transcripts encode the expression of the second, third, and fourth enzymes in the pathway, 5-aminolevulinate dehydratase (ALA-dehydratase), hydroxymethylbilane synthase (HMB-synthase), and URO-synthase, although only ALAS and HMB-synthase (HMBS) genes encode unique housekeeping and erythroid-specific isozymes. The characteristics of the heme biosynthetic genes and their enzymes are summarized in Table 1. All 9 human genes have been cloned, their structures determined or predicted, many of their porphyria-causing mutations identified, and murine models generated or naturally occurring animal counterparts identified.1,2,5 As shown in Figure 1, pathway intermediates are the porphyrin precursors, ALA and porphobilinogen (PBG), and porphyrins (mostly in their reduced forms, known as porphyrinogens). In humans, these intermediates do not accumulate in significant amounts under normal conditions nor do they have important physiologic functions.
The pathway is under tight regulatory control, particularly in the liver, where heme biosynthesis is modulated by feedback control of the first and rate-limiting enzyme in the pathway, ALAS1. Erythroid heme biosynthesis is regulated by the availability of iron, which regulates translation of the erythroid-specific ALAS2 mRNA and serves as a substrate for the introduction of iron into protoporphyrin by ferrochelatase (FECH). In the liver, “free” heme regulates the synthesis and mitochondrial translocation of the housekeeping form of ALA-synthase, ALAS1.6 Heme represses the synthesis of the ALAS1 mRNA and blocks the transport of the enzyme from the cytosol into mitochondria. ALAS1 can be induced by a variety of drugs, steroids, nutrition, stress, and other chemicals, which can trigger the acute neuropathic attacks in the acute hepatic porphyrias. Hepatic ALAS1 can be induced by many of the same chemicals that induce cytochrome P450 enzymes in the endoplasmic reticulum of the liver. The other hepatic heme biosynthetic enzymes are presumably expressed at constant levels, although their relative activities and kinetic properties differ. To date, no mutations or functional polymorphisms have been identified in ALAS1.
In contrast, in the erythron, regulatory mechanisms allow for the production of the very large amounts of heme needed for hemoglobin synthesis. The erythroid-specific ALAS2 gene is expressed at ∼ 30-fold higher levels than the hepatic enzyme, and an erythroid-specific control mechanism regulates iron transport into erythroid cells. Translation of the ALAS2 mRNA is modulated by the binding of the iron regulatory proteins to an iron response element in the 5′ region of the ALAS2 mRNA. Translation is inhibited when an iron regulatory protein is bound to the iron response element, and the binding activity is regulated by iron. Translation is down-regulated when iron is deficient and up-regulated when iron is abundant. During erythroid differentiation, the activities of other heme biosynthetic enzymes may be increased. Loss-of-function mutations in the erythroid ALAS2 cause X-linked gene sideroblastic anemia,1,7 whereas gain-of-function mutations in the last exon of ALAS2 cause the recently recognized X-linked form of erythropoietic protoporphyria (EPP), XLP.8
Classification of the porphyrias
Traditionally, the porphyrias have been classified as either hepatic or erythropoietic depending on the primary site of overproduction and accumulation of porphyrin precursors or porphyrins, although some porphyrias have overlapping features.1,4 The hepatic porphyrias are characterized by overproduction and initial accumulation of the porphyrin precursors, ALA and PBG, and/or porphyrins primarily in the liver, whereas in the erythropoietic porphyrias, overproduction and initial accumulation of the pathway intermediates occur primarily in bone marrow erythroid cells. Here, for simplicity, we have classified the 8 major porphyrias into 3 groups: (1) the 4 acute hepatic porphyrias, (2) the single hepatic cutaneous porphyria PCT, and (3) the 3 erythropoietic cutaneous porphyrias (Table 1). It should be appreciated that there is some overlap, as patients with the acute hepatic porphyrias, hereditary coproporphyria (HCP), and variegate porphyria (VP) may have cutaneous lesions, and the rare homozygous dominant forms of acute intermittent porphyria (AIP), HCP, and VP, as well as the rare homozygous recessive form of PCT, hepatoerythropoietic porphyria, have erythropoietic manifestations.9–12
The acute hepatic porphyrias
Clinical manifestations
This group includes 3 autosomal dominant porphyrias, AIP, HCP, VP, and autosomal recessive ALA-dehydratase-deficient porphyria (ADP; Table 2). Of these, AIP is the most common and ADP is rare with < 10 patients reported to date. The major manifestations of these disorders are the life-threatening acute neurologic attacks, which typically occur after puberty. Abdominal pain, the most common symptom, is usually steady and poorly localized but may be cramping. Constipation, abdominal distention, and decreased bowel sounds are common. Increased bowel sounds and diarrhea are less common. Because inflammation is absent, abdominal tenderness, fever, and leukocytosis are usually not prominent. Additional common manifestations include nausea; vomiting; tachycardia; hypertension; mental symptoms; extremity, neck, or chest pain; headache; muscle weakness; sensory loss; tremors; sweating; dysuria; and bladder distention.
Human porphyrias: major clinical and laboratory features

AR indicates autosomal recessive; AD, autosomal dominant; XL, X-linked; NV, neurovisceral; CP, cutaneous photosensitivity; and —, not applicable.
*Increases that may be important for diagnosis.
†A polymorphism in intron 3 of the wild-type allele affects the level of enzyme activity and clinical expression.
‡Increased activity resulting from “gain-of-function” mutations in ALAS2 exon 11.
Diagnosis
During acute attacks, the urinary porphyrin precursor PBG is virtually always markedly increased in AIP, HCP, and VP and is not in any other medical condition. The Trace PBG Kit (Trace America/Trace Diagnostics) is a rapid diagnostic method to detect elevated levels of urinary PBG. The same spot urine specimen should be saved to quantitatively determine ALA and PBG levels to confirm the qualitative PBG result and to detect elevations of ALA in rare patients with ADP. Subsequent urinary (and fecal) porphyrin analyses can suggest a specific acute hepatic porphyria (Table 2). Once a biochemical diagnosis is established, mutation analysis of the genes for AIP (HMBS), HCP (CPOX), VP (PPOX), and then ADP (ALAD) should be undertaken. Molecular diagnostic studies also are useful to confirm the diagnosis of patients who had attacks and to identify at-risk family members once the specific mutation is known in the index case. To date, more than 375 mutations for AIP, more than 60 for HCP, and more than 165 for VP have been identified; ADP is rare and only 12 ALAD mutations have been detected.13 Once the causative mutation is identified in the index case, it is important to identify at-risk relatives and to offer asymptomatic heterozygote counseling to avoid the drugs, fasting, hormones, and other precipitants of the acute attacks.
To date, there have been no genotype/phenotype correlations in the acute hepatic porphyrias, with the exception of severely affected children with homozygous dominant AIP,11 VP,10 and HCP and its mutation-specific variant form, harderoporphyria.9,12 In addition, there have been no genome wide association studies or exomic/genomic sequencing efforts to identify the modifying or predisposing genes in the patients who have chronic or frequent attacks. Mutation analysis is available for diagnosis of each porphyria (Mount Sinai Porphyria Diagnostic Laboratory; 866-322-7963).
Pathogenesis of the acute attacks
For years, the pathogenesis of these attacks was intensely debated.14,15 Did they result from: (1) elevated levels of ALA and PBG, one or both being neurotoxic, (2) heme deficiency in the nervous system, or (3) both? Clearly, induction of the acute attacks is related to environmental or hormonal factors, such as drugs, diet, and steroid hormones that induce ALAS1 resulting in the overproduction of ALA and PBG.
Recently, new insight into the etiology of the acute attacks came from several patients with AIP and VP who had chronic attacks that ceased after liver transplantation.16,17 Of note, “domino” transplantation of the “porphyric livers” into recipients with liver failure who did not qualify for transplantations, corrected their liver function, but made them susceptible to acute attacks. Thus, the debate was resolved. The acute attacks result from the hepatic production of a neurotoxic substance, presumably ALA (a γ-aminobutyric acid analog) and/or PBG that may interact with γ-aminobutyric acid or glutamate receptors.
Treatment of acute attacks
Consensus guidelines for the treatment of the acute attacks of the acute hepatic porphyrias are available.3 Briefly, during acute attacks, narcotic analgesics are usually required for abdominal pain, and phenothiazines are effective for nausea, vomiting, anxiety, and restlessness. Insomnia and restlessness are treated with chloral hydrate or low doses of certain short-acting benzodiazepines. Carbohydrate loading, usually with intravenous glucose (at least 300 g/day), may be effective in milder acute attacks (without paresis, hyponatremia, etc). Because intravenous hemin is more effective and the response slower if treatment is delayed, it is recommended that hemin therapy be started initially for moderate to severe attacks, and for mild attacks that do not respond to carbohydrate loading within 1-2 days. The standard regimen is 3-4 mg of heme in the form of lyophilized hematin (Panhematin, Lundbeck Pharmaceuticals), heme albumin (hematin reconstituted with human albumin), or heme arginate (Orphan Europe), infused daily for 4 days.3 That increased carbohydrates may ameliorate attacks is supported by the recent finding that hepatic ALAS1 is modulated by the peroxisome proliferator-activated receptor-γ coactivator 1α, providing an important link between nutritional status and the acute attacks.18
Allogeneic liver transplantation has been performed in AIP and VP patients who had chronic or frequent acute attacks that were not responsive to treatment.16,17 After transplantation, their elevated urinary ALA and PBG levels returned to normal in 24 hours, and the recipients have not experienced acute attacks for years since transplantation. However, liver transplantation is a high-risk procedure and should be considered only as a last resort in patients with severe recurrent attacks.19 Recently, liver-directed gene therapy has been successful in the prevention of drug-induced biochemical attacks in a murine model of human AIP.20
The hepatic cutaneous porphyria: PCT
PCT, the most common of the porphyrias, may be either sporadic (type 1) or familial (type 2). For clinical symptoms to manifest in either type, the hepatic URO-decarboxylase activity must be 20% of normal or less. Type 1 (or sporadic) PCT patients have no URO-decarboxylase (UROD) mutations and, when asymptomatic, have normal URO-decarboxylase activity systemically. Type 2 (or familial) PCT patients are heterozygous for UROD mutations, and asymptomatic patients have approximately half-normal enzyme activity systemically. Of note, although type 2 PCT is an autosomal dominant disease, it is incompletely penetrant; the half-normal enzyme activity in type 2 patients is a significant predisposing factor but is insufficient by itself to cause symptomatic PCT. Other genetic and environmental factors contribute to susceptibility in both types 1 and 2 PCT. For this reason, penetrance of this genetic trait is low, and many patients who present with type 2 PCT have no family history of the disease and may appear to have sporadic disease. Although a diagnosis of PCT is usually made by a family physician, internist, and/or dermatologist the most common treatment for this disease is repetitive phlebotomies, so these patients are typically referred to hematologists. Phlebotomies are thought to decrease the hepatic iron load. An alternative oral treatment for PCT is low dose chloroquine treatment (see “Treatment”).
Clinical manifestations
PCT usually presents in adults and is characterized by blistering skin lesions that appear most commonly on the backs of the hands (Figure 2). These rupture and crust over, leaving areas of atrophy and scarring. Lesions may also occur on the forearms, face, legs, and feet. Skin friability and small white papules termed milia are common, especially on the backs of the hands and fingers. Hypertrichosis and hyperpigmentation, particularly on the face, are especially troublesome in women. Occasionally, the skin in sun-exposed areas becomes severely thickened with scarring and calcification, which resembles systemic sclerosis. Neurologic features are absent.

Porphyria cutanea tarda. (A-B) Sun-exposed hands of a PCT patient showing areas of atrophy and scarring. (C) Urine from a symptomatic PCT patient and a healthy control in daylight (left) and under ultraviolet light (right). The PCT urine has an orange-red color in daylight that fluoresces red under ultraviolet light.
Porphyria cutanea tarda. (A-B) Sun-exposed hands of a PCT patient showing areas of atrophy and scarring. (C) Urine from a symptomatic PCT patient and a healthy control in daylight (left) and under ultraviolet light (right). The PCT urine has an orange-red color in daylight that fluoresces red under ultraviolet light.
Diagnosis
The urinary ALA level may be slightly increased, but the PBG level is normal. Urinary porphyrins are highly elevated and consist mostly of uroporphyrin and heptacarboxylate porphyrin, which is a diagnostic pattern for PCT and its rare homozygous variant, hepatoerythropoietic porphyria. Plasma porphyrins are also increased, which is useful for screening. Increased isocoproporphyrin, primarily in feces, is diagnostic for URO-decarboxylase deficiency.1
URO-decarboxylase activity in erythrocytes is generally about half-normal in type 2 PCT and normal in type 1 PCT. In hepatoerythropoietic porphyria, the enzyme activity is ∼ 3%-10% of normal systemically. UROD gene mutation analysis is recommended for diagnosis as even sporadic patients with no family history may have predisposing UROD mutations, reclassifying them as type 2 PCT. To date, > 105 UROD mutations have been identified.13
Pathogenesis
When the cutaneous lesions are present in both types 1 and 2 patients, the hepatic URO-decarboxylase activity is reduced to < 20% of normal. Thus, investigators searched for an inhibitor that reduced the hepatic enzyme activity in symptomatic type 1 and 2 patients. Recently, uroporphomethene, an oxidized form of uroporphyrinogen, the substrate of URO-decarboxylase, was shown to be the enzyme inhibitor.21 Of note, the oxidation of uroporphyrinogen to the inhibitor is iron dependent in the liver, emphasizing the importance of iron overload as a causative factor and therapeutic target.
Multiple susceptibility factors, in addition to inherited UROD mutations in type 2 PCT, appear to act synergistically to cause the oxidative stress and iron overload needed to generate the inhibitor and cause clinical expression.22–24 Chronic hepatitis C virus, HIV, alcohol abuse, and estrogen use in women are all contributing risk factors.23,25 The importance of excess hepatic iron is underscored by the increased prevalence of the common hemochromatosis (HFE)-causing mutations, C282Y and H63D, in patients with types 1 and 2 PCT. However, the iron overload usually is mild or moderate in degree. Notably, chronic hepatitis C virus and alcohol abuse decrease hepcidin production by hepatocytes, leading to increased intestinal iron absorption. The increased hepatic iron and oxidative stress leads to the formation of the enzyme inhibitor and oxidation of porphyrinogens to porphyrins.21,24
Treatment
Discontinuing risk factors, such as alcohol, estrogens, and iron supplements, is recommended but may not result in timely improvement. A complete response can almost always be achieved by repeated phlebotomy to reduce hepatic iron. A unit of blood can be removed approximately every 2 weeks. The aim is to gradually reduce iron until the serum ferritin reaches the lower limits of normal (< 25 ng/mL).26 Hemoglobin levels or the hematocrit should be followed closely to prevent anemia. Because iron overload is not marked in most cases, the target ferritin level can often be achieved after only 5 or 6 phlebotomies; however, PCT patients with hemochromatosis may require many more phlebotomies. To document improvement in PCT, one can follow the total plasma porphyrin concentration, which becomes normal after the target ferritin level is reached. After remission, continued phlebotomy may not be needed. Alternatively, a low-dose regimen of chloroquine or hydroxychloroquine (eg, 125 mg chloroquine phosphate twice weekly), which mobilizes excess porphyrins from the liver and promotes their excretion, is a useful alternative to phlebotomy, especially when phlebotomy is contraindicated or poorly tolerated. Hepatic imaging can detect or exclude complicating hepatocellular carcinoma. Phlebotomy treatment for PCT in patients with end-stage renal disease is facilitated by administration of erythropoietin.
The erythropoietic cutaneous porphyrias
The erythropoietic cutaneous porphyrias, congenital erythropoietic porphyria (CEP) and EPP, and its recently recognized X-linked form, XLP, are characterized by elevations of porphyrins in bone marrow and erythrocytes and usually present with cutaneous photosensitivity in infancy or early childhood,1,8 or in the case of CEP, even in utero as nonimmune hydrops fetalis.27
Congenital erythropoietic porphyria
CEP is an autosomal recessive disorder that results from the markedly deficient, but not absent, activity of URO-synthase and the resultant accumulation of uroporphyrin I and coproporphyrin I isomers1,27 (Table 2). Uroporphyrinogen I, which is derived from the nonenzymatic cyclization of the substrate HMB, is metabolized by URO-decarboxylase to coproporphyrinogen I, but the latter is not a substrate for COPRO-oxidase. CEP is associated with hemolytic anemia and severe cutaneous photosensitivity. Excess porphyrins are also deposited in teeth and bones.
Clinical manifestations
Severe cutaneous photosensitivity begins in early infancy in most cases.27 The disease may be recognized in utero as a cause of nonimmune hydrops fetalis. The skin over light-exposed areas is friable, and bullae and vesicles are prone to rupture and infection (Figure 3). Skin thickening, focal hypopigmentation and hyperpigmentation, and hypertrichosis of the face and extremities are characteristic. Secondary infection and bone resorption may lead to disfigurement of the face and hands. The teeth are reddish brown and fluoresce on exposure to long-wave ultraviolet light. Hemolysis is probably the result of the marked increase in erythrocyte porphyrins and leads to splenomegaly. A later-onset, milder form of the disease has been reported.28,29 However, several of these later-onset patients had a myeloid malignancy (usually myelodysplastic disorder) and were found to have normal erythrocyte URO-synthase activity, and no germline URO-synthase (UROS) or GATA1 mutations; this entity was termed erythropoietic uroporphyria, presumably because only a minor clone of uroporphyric cells had somatic UROS mutations causing the uroporphyrinogen I accumulation and the late-onset phenotype.30

Congenital erythropoietic porphyria. (A) A severely affected CEP patient who has had multiple sun-induced skin lesions. The cutaneous bullae and vesicles burst and became secondarily infected, leading to bone involvement and resultant loss of facial features and digits. (B) Note his brownish discolored teeth, which fluoresce (erythrodontia) when exposed to ultraviolet light. The erythrodontia is the result of the accumulation of uroporphyrin I and coproporphyrin I in his teeth. (C) Urine from a CEP patient that fluoresces red under ultraviolet light (left) and from a healthy person (right).
Congenital erythropoietic porphyria. (A) A severely affected CEP patient who has had multiple sun-induced skin lesions. The cutaneous bullae and vesicles burst and became secondarily infected, leading to bone involvement and resultant loss of facial features and digits. (B) Note his brownish discolored teeth, which fluoresce (erythrodontia) when exposed to ultraviolet light. The erythrodontia is the result of the accumulation of uroporphyrin I and coproporphyrin I in his teeth. (C) Urine from a CEP patient that fluoresces red under ultraviolet light (left) and from a healthy person (right).
Diagnosis
Uroporphyrin and coproporphyrin (mostly type I isomers) accumulate in the bone marrow, circulating erythrocytes, plasma, urine, and feces. The diagnosis is confirmed by the demonstration of markedly deficient URO-synthase activity or the identification of specific mutations in the UROS gene. The disease can be detected in utero by measuring porphyrins in amniotic fluid and URO-synthase activity in cultured amniotic cells or chorionic villi, or by the detection of the family's specific UROS mutations. To date, > 35 UROS mutations have been identified, including 4 in its erythroid-specific promoter, and genotype/phenotype correlations have identified patients with severe and milder manifestations.13,27,31 An X-linked variant of CEP caused by a mutation in GATA1, an X-linked transcription factor that binds to globin gene and heme biosynthetic gene promoters and enhancers, has been reported.32 More recently, a severely affected CEP patient with 2 UROS mutations was found to have a gain-of-function mutation in the ALAS2 gene, which increased disease severity compared with affected siblings who did not have the ALAS2 mutation.33
Treatment
Severe cases often require chronic transfusions for anemia, which can be started in utero. Chronic erythrocyte transfusions sufficient to suppress erythropoiesis are effective in reducing porphyrin production but result in iron overload and other complications.34 Protection from sunlight is essential, and minor skin trauma should be avoided. Complicating bacterial infections should be treated promptly. Bone marrow and cord blood transplantation have proved effective in several transfusion-dependent children,32,35 providing the rationale for stem-cell gene therapy.27,36 Recently, efforts to rescue the common UROS mutation (C73R) with a pharmacologic chaperone and/or protease inhibitor were reported.37
EPP and XLP
EPP is an autosomal recessive disorder resulting from mutations in the FECH gene that markedly reduce the activity of FECH, the last enzyme in the heme biosynthetic pathway. EPP is the most common erythropoietic porphyria and the most common porphyria in children.1 Recently, a clinically indistinguishable X-linked form of EPP, XLP, was identified that results from gain-of-function mutations in the last exon of the ALAS2 gene that prematurely truncate the carboxyl-terminus of the encoded erythroid-specific ALAS2 enzyme and increase its activity.8
Clinical manifestations
Skin photosensitivity, which differs from that in other porphyrias, usually begins in early childhood and consists of pain, redness, and itching occurring within minutes of sunlight exposure (Figure 4). Vesicular lesions are uncommon. The burning pain that develops shortly after sun exposure can be excruciating, and not relieved by narcotic analgesics. Subsequently, the exposed area becomes red and swollen. Vesicles and bullae are sparse and occur in only ∼ 10% of cases. Chronic skin changes may include lichenification, leathery pseudovesicles, labial grooving, and nail changes. Severe scarring is rare, as are pigment changes, friability, and hirsutism. Hemolysis and anemia are usually absent or mild. Patients also have been described with palmar keratosis.38 In addition, later-onset EPP patients associated with myleoplastic syndromes have been described.39 EPP and XLP patients may have liver disease that may cause minor abnormalities of liver function or result in liver failure. Protoporphyric liver disease may cause severe abdominal pain, especially in the right upper quadrant, and back pain. Gallstones composed at least in part of protoporphyrin may be symptomatic in EPP patients, especially in children, and need to be excluded as a cause of biliary obstruction in patients with hepatic decompensation.40 However, in ∼ 5% of the patients, the accumulation of protoporphyrin causes liver disease that may be chronic but sometimes develops rapidly and may progress to liver failure and death.

Erythropoietic protoporphyria. (A) An EPP patient after sun exposure. Note the reddish and swollen appearance of her face and (B) scarring and thickening of the skin on the dorsum of her hand because of multiple sun/light exposures.
Erythropoietic protoporphyria. (A) An EPP patient after sun exposure. Note the reddish and swollen appearance of her face and (B) scarring and thickening of the skin on the dorsum of her hand because of multiple sun/light exposures.
Diagnosis
The primary source of excess protoporphyrin in EPP and XLP is the bone marrow reticulocyte, and erythroid cells exhibit red fluorescence when examined by fluorescence emission microscopy at 620 nm with excitation at 405 nm. The diagnosis of EPP and XLP is made by demonstrating markedly increased erythrocyte protoporphyrins. In EPP, erythrocyte protoporphyrin is almost all free (not complexed with zinc) and is mostly bound to hemoglobin. In XLP, free and zinc protoporphyrin are both increased in erythrocytes.8 Urinary levels of porphyrins and porphyrin precursors are normal. The biochemical diagnosis is best confirmed by mutation analyses of the FECH and ALAS2 genes. To date, > 135 FECH loss-of-function mutations have been identified, including a common low expression allele (IVS3–48T > C), which is present in ∼ 10% of European whites with various frequencies in other ethnic/racial groups.41 Many loss-of-function mutations result in an unstable or absent enzyme protein. The common IVS3–48T > C splicing mutation results in ∼ 25% normal FECH transcripts. Most EPP patients (∼ 90%) have a FECH loss-of-function mutation in cis and the common low expression allele in trans, resulting in 15%-25% of normal FECH activity. In ∼ 5%-10% of EPP families, 2 FECH loss-of-function mutations have been found.25,41,42 These patients may have a variant EPP phenotype with palmar keratosis.38 The 2 exon 11 ALAS2 mutations that cause XLP alter the carboxyl-terminal amino acid sequence and result in increased ALAS2 activity and the accumulation of free and zinc-protoporphyrins.8 In Western Europe, XLP accounts for less than 5% of cases with the EPP phenotype.42
Pathogenesis
Protoporphyrin accumulates primarily in bone marrow reticulocytes during hemoglobin synthesis, and then appears in plasma, is taken up in the liver, and is excreted in bile and feces. Protoporphyrin taken up by the skin and its blood vessels are photoactivated by sun/light, resulting in the cutaneous photosensitivity. The mechanism responsible for the protoporphyrin accumulation, even in the presence of adequate iron, is not clear and is the focus of current research.
Protoporphyrin is insoluble at neutral pH, and excess amounts form crystalline structures in liver cells and can decrease hepatic bile flow in bile fistula rats. Rapidly progressive liver disease in human EPP is associated with increasing protoporphyrin levels in liver, plasma, and erythrocytes and increased photosensitivity. Hepatic complications appear to be higher in autosomal recessive EPP because of 2 FECH mutations and in XLP.43
Treatment
Avoiding sunlight exposure and wearing clothing designed to provide protection for conditions with chronic photosensitivity are essential. Oral β-carotene (120-180 mg/dL), which causes a mild skin discoloration because of carotenemia, may improve tolerance to sunlight, but the beneficial effects may be limited. Recently, clinical studies have shown that treatment with an α-melanocyte-stimulating hormone analog, which darkens the skin, can increase tolerance to sunlight exposure without pain.44
Treatment of hepatic complications is difficult. Cholestyramine and other porphyrin absorbents, such as activated charcoal, may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement. Plasmapheresis and intravenous hemin are sometimes beneficial. However, liver transplantation may be necessary and is often successful in the short term, but liver disease eventually reoccurs in the transplanted liver because of continued bone marrow production of excess protoporphyrin (for review, see McGuire et al45 ). Post-transplantation treatment with hemin and plasmapheresis may help prevent recurrence. However, bone marrow transplantation, which has been successful in human EPP and prevented liver disease in a mouse model,46 should be considered along with or after liver transplantation.
In conclusion, the inborn errors of heme biosynthesis, the porphyrias, are a diverse group of metabolic disorders that often are diagnosed and treated by hematologists. Recent advances in our understanding of these disorders include the recognition of their genetic heterogeneity and disease variants, improved molecular diagnostics, genotype/phenotype correlations, and current and experimental treatments. Current research is focused on developing gene and stem cell replacement, pharmacologic chaperones to rescue misfolded mutant enzymes, and future efforts to correct specific mutations in induced pluripotent stem cells from porphyria patients followed by transplantation of hepatic or erythroid derived and corrected cells. The recent and future advances should markedly improve our diagnosis and management of these diseases.
This article was selected by the Blood and Hematology 2012 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2012. This article is reprinted with permission from Blood. 2012; Volume 120.
Acknowledgments
The authors thank Ms Nicole Kelly for her assistance in the preparation of the manuscript and Dr David Bishop for his critical review and helpful suggestions.
This work was supported in part by the National Institutes of Health, including the Rare Disease Clinical Research Network for the Porphyrias Consortium (grant U54 DK083909 and research grant 5 R01 DK026824).
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: R.J.D. was the Principal Investigator of a Phase 2 multi-center, randomized, placebo-controlled, double-blinded study evaluating the safety and efficacy of Afamelanotide (aalpha-melanocyte stimulating hormone) for the treatment of erythropoietic protoporphyria. The sponsor was Clinuvel Pharmaceuticals Limited. R.J.D. has no financial relationship with the company.
Correspondence
Robert J. Desnick, Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, Box 1498, Fifth Ave and 100th St, New York, NY 10029-6574; Phone: 212-659-6700; Fax: 212-360-1809; e-mail: robert.desnick@mssm.edu.
